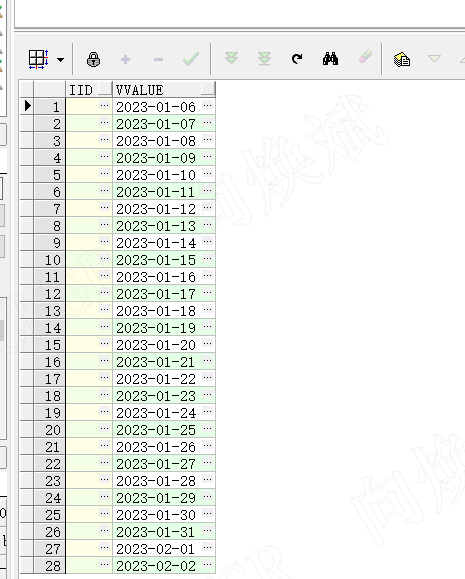
蛋白质是一个较长的氨基酸序列,比如100个氨基酸的规模,如此长的氨基酸序列连在一起是不稳定的,它们会卷在一起,形成一个独特的3D结构,这个3D结构的形状决定了蛋白质的功能。
蛋白质结构预测(蛋白质折叠问题):输入氨基酸序列,预测3D结构。
目前,我们知道10万种蛋白质的结构,但是我们已知的蛋白质有10亿种,因此,我们只了解这10万种蛋白质的功能(3D结构),用传统观测方法(冷冻电镜:将蛋白质冷冻,用电子显微镜从各角度观测,然后还原3D结构),我们需要数月数年才能了解一个蛋白质的结构。
AlphaFold 1的预测精度没有达到原子级别:对某个氨基酸位置的预测和实验测量的差距不在一个原子级别大小内。AlphaFold 2在CASP14(科学家将每两年内实验测量的新蛋白质结构用于该比赛的评估)中出现,并达到了原子级别。

- a:竞赛方法对比,AlphaFold 2具有更低的偏差。b:模型预测与实验观测的对比。c:用碳原子进行比较,碳原子的大小是1.5A。d:大型蛋白质的预测和实验对比。
AlphaFold 2的架构如下:

- 输入为人类蛋白质的氨基酸序列,序列中的每个点(正方形,圆形,三角形)表示一个氨基酸。输出为预测的每个氨基酸在3D空间中的位置。
AlphaFold 2分为3个部分:特征提取,编码器,解码器。
对于特征提取,包括:
- 序列的特征:首先人类的序列被直接copy,同时进入genetic database搜索类似的序列(寻找有相似片段的蛋白质),比如找到了来自鱼类,兔子,鸡的序列。我们将它们拿出来作为MSA(multiple sequence alignments,多序列比对,两序列尽可能多地对齐相同氨基酸)。
- 氨基酸之间的特征:另一个特征是氨基酸之间的关系,我们知道,蛋白质折叠是氨基酸原子之间的相互作用,因此,我们在pairing中显式表示每两个氨基酸之间的关系。比如图中蛋白质长度为6,因此得到6×6的表,最开始,表中元素可以是序列视角上的距离,我们希望通过网络推理最终得到3D空间中的氨基酸之间的距离。同时,我们进入structure database(现有已知的蛋白质结构)进行搜索,得到很多个可能的距离模板。
对于编码器,输入为两个3D的张量:
- 对于MSA张量,s代表序列的数量,包括s-1个相似的序列。r表示每个蛋白质序列包含r个氨基酸。c则表示每个氨基酸被表示为长度为c的向量。
- 对于pair张量,则为 r×r 的表,每个元素是长度为c的向量,作为氨基酸之间的距离embedding。
- 两个张量输入Transformer变种Evoformer,Transformer是处理序列之间的关系,不能处理2D之间的关系,另外由于输入的是两个不同的张量,也不能用Transformer处理。
编码器经过信息流动,切片输出人类氨基酸的序列编码,形状为r×c,以及pair的表示。
解码器基于编码器的两个输出,得到3D结构信息。AlphaFold 2还包括回收机制,将编码器输出和解码器输出回收到编码器输入。注意回收机制不是循环神经网络,循环神经网络是允许梯度在回收路线上传播的,但AlphaFold不允许梯度在回收路线上传播。
下面详细了解编码器的架构,下面是Evoformer中一个block的结构:

MSA序列信息首先被按行处理(对应s),然后按列处理(对应r),Transition用MLP实现非线性变换。Outer product mean实现MSA与pair的信息融合。
对于解码器,首先了解蛋白质的3D结构表达。

最简单的方式就是将每个原子在3D空间中的坐标记录下来,也就是三个值,我们只要对每个原子预测三个值就行。但是我们要知道,我们将蛋白质在3D空间中旋转,位移都不会影响其结构,但会改变绝对位置中坐标的值。因此,我们需要使用相对位置。如上图所示,蛋白质实际上是氨基酸连接的序列,我们只要记录上一个氨基酸相对下一个氨基酸的位置(旋转角度,相对距离),根据每个氨基酸的相对位置,我们就能还原出蛋白质的3D结构。

解码器首先预测主干的形状(backbone frame),然后预测主干上枝叶的旋转和距离,得到氨基酸序列的3D结构。
训练

训练的主要损失函数为FAPE,根据预测出的刚体变换信息,可以还原蛋白质的3D结构,图中绿色为预测出的结构,灰色为真实结构。将预测原子位置(图中
x
i
x_{i}
xi)和真实原子位置相减得到距离。距离越大,FAPE越大。
另外还有一个训练策略(noisy student),模型先在PDB上有监督学习,然后用于更大的无标签数据上推理,取出预测置信度高的结构加入噪声(比如数据增强)作为新的有标签数据,从而扩大训练数据的规模。