GROMACS 教程–水中的溶菌酶
中文教程:http://jerkwin.github.io/
英文教程:http://www.mdtutorials.com
此示例将指导新用户完成模拟系统的设置过程,该模拟系统在一盒水和离子中包含蛋白质(溶菌酶)。每个步骤都将包含输入和输出的说明,使用一般设置的典型设置。
一些 GROMACS 基础知识
随着 GROMACS 5.0 版的发布,所有工具基本上都是名为“gmx”的二进制文件的模块。这与以前的版本不同,在以前的版本中,每个工具都作为自己的命令被调用。在 5.0 中,这仍然通过符号链接起作用,但它们会在未来的版本中消失,所以最好习惯新的做事方式。要获取有关任何 GROMACS 模块的帮助信息,您可以调用以下任一命令:
gmx help (module)
或者
gmx (module) -h
其中 (module) 替换为您要发出的命令的实际名称。信息将打印到终端,包括可用的算法、选项、所需的文件格式、已知的错误和限制等。对于 GROMACS 的新用户,调用常用命令的帮助信息是了解每个命令可以做什么的好方法. 现在,开始有趣的事情!
溶菌酶教程

我们必须下载我们将要使用的蛋白质结构文件。在本教程中,我们将使用鸡蛋清溶菌酶(PDB 代码 1AKI)。转到RCSB网站并下载晶体结构的 PDB 文本。
下载结构后,您可以使用 VMD、Chimera、PyMOL 等查看程序可视化结构。查看分子后,您会想要去除结晶水。要删除水分子(PDB 文件中的残留“HOH”),请使用纯文本编辑器,如 vi、emacs (Linux/Mac) 或记事本 (Windows)。不要使用文字处理软件!或者,您可以使用grep非常轻松地删除这些行:
grep -v HOH 1aki.pdb > 1AKI_clean.pdb
请注意,这样的程序并不是普遍适用的(例如,紧密结合或其他功能性活性位点水分子的情况)。对于我们的意图,我们不需要结晶水。
始终检查您的 .pdb 文件中是否有注释 MISSING 下列出的条目,因为这些条目表示晶体结构中不存在的原子或整个残基。终端区域可能不存在,并且可能不会出现动态问题。不完整的内部序列或任何缺少原子的氨基酸残基都会导致 pdb2gmx 失败。这些缺失的原子/残基必须使用其他软件包进行建模。*另请注意,pdb2gmx 并不神奇。它不能为任意分子生成拓扑结构,只能为力场定义的残基(在 .rtp 文件中——通常是蛋白质、核酸和数量非常有限的辅因子,如 NAD(H) 和 ATP)。
现在结晶水已经消失并且我们已经验证所有必需的原子都存在,PDB 文件应该只包含蛋白质原子,并准备好输入到第一个 GROMACS 模块 pdb2gmx。pdb2gmx 的目的是生成三个文件:
- 分子的拓扑结构。
- 位置限制文件。
- 后处理结构文件。
拓扑(默认为 topol.top)包含在模拟中定义分子所需的所有信息。此信息包括非键参数(原子类型和电荷)以及键参数(键、角度和二面角)。生成拓扑后,我们将对其进行更详细的查看。
通过发出以下命令来执行 pdb2gmx:
gmx pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce
该结构将由 pdb2gmx 处理,系统将提示您选择一个力场:
选择力场:
来自“/usr/local/gromacs/share/gromacs/top”:
1:AMBER03蛋白,核酸AMBER94(Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2:AMBER94 力场(Cornell et al., JACS 117, 5179-5197, 1995)
3:AMBER96 蛋白、核酸 AMBER94(Kollman 等人,Acc. Chem. Res. 29, 461-469, 1996)
4:AMBER99 蛋白、核酸 AMBER94(Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5:AMBER99SB 蛋白,核酸 AMBER94(Hornak 等人,Proteins 65, 712-725, 2006)
6:AMBER99SB-ILDN 蛋白,核酸 AMBER94(Lindorff-Larsen 等人,Proteins 78, 1950-58, 2010)
7:AMBERGS 力场 (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8:CHARMM27 全原子力场(CHARM22 加 CMAP 用于蛋白质)
9:GROMOS96 43a1力场
10:GROMOS96 43a2力场(改进的烷烃二面角)
11:GROMOS96 45a3力场(舒勒JCC 2001 22 1205)
12: GROMOS96 53a5 力场 (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 力场 (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 力场 (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15:OPLS-AA/L 全原子力场(2001 氨基酸二面角)
力场将包含将写入拓扑的信息。这是一个非常重要的选择!您应该始终通读每个力场并决定哪个最适合您的情况。对于本教程,我们将使用全原子 OPLS 力场,因此在命令提示符下键入 15,然后按“Enter”。
还有许多其他选项可以传递给 pdb2gmx。这里列出了一些常用的:
-ignh:忽略PDB文件中的H原子;对 NMR 结构特别有用。否则,如果存在 H 原子,它们的名称必须与 GROMACS 中的力场预期的完全相同。存在不同的约定,因此处理 H 原子有时会让人头疼!如果您需要保留初始 H 坐标,但需要重命名,那么 Linux sed 命令是您的好帮手。-ter:交互分配 N- 和 C- 末端的电荷状态。-inter:交互式分配 Glu、Asp、Lys、Arg 和 His 的电荷状态;选择哪些半胱氨酸参与二硫键。
您现在已经生成了三个新文件:1AKI_processed.gro、topol.top 和 posre.itp。1AKI_processed.gro 是一个 GROMACS 格式的结构文件,其中包含力场内定义的所有原子(即,H 原子已添加到蛋白质中的氨基酸中)。topol.top 文件是系统拓扑(稍后会详细介绍)。posre.itp 文件包含用于限制重原子位置的信息(稍后会详细介绍)。
最后一点:许多用户认为 .gro 文件是必需的。 这不是真的。GROMACS 可以处理许多不同的文件格式,.gro 只是写入坐标文件的命令的默认格式。它是一种非常紧凑的格式,但精度有限。例如,如果您更喜欢使用 PDB 格式,您需要做的就是指定一个带有 .pdb 扩展名的适当文件名作为输出。pdb2gmx 的目的是产生一个符合力场的拓扑;输出结构主要是此目的的副作用,旨在方便用户。格式可以是您喜欢的任何格式(有关不同格式,请参阅 GROMACS 手册)。
让我们看看输出拓扑 (topol.top) 中的内容。再次使用纯文本编辑器检查其内容。在几个注释行(前面有 ;)之后,您会发现以下内容:
#include "oplsaa.ff/forcefield.itp"
此行调用 OPLS-AA 力场中的参数。它位于文件的开头,表明所有后续参数均源自此力场。下一个重要的行是[ moleculetype ],在它下面你会发现
; Name nrexcl
Protein_A 3
“蛋白质A”这个名字定义了分子的名称,基于蛋白质在PDB文件中被标记为链A的事实。对于绑定邻居有3个例外。关于排除的更多信息可以在GROMACS手册中找到;关于这些信息的讨论超出了本教程的范围。
下一节定义[ atoms ]蛋白质中的。信息显示为列:
[ atoms ]
; nr type resnr residue atom cgnr charge mass typeB chargeB massB
; residue 1 LYS rtp LYSH q +2.0
1 opls_287 1 LYS N 1 -0.3 14.0067 ; qtot -0.3
2 opls_290 1 LYS H1 1 0.33 1.008 ; qtot 0.03
3 opls_290 1 LYS H2 1 0.33 1.008 ; qtot 0.36
4 opls_290 1 LYS H3 1 0.33 1.008 ; qtot 0.69
5 opls_293B 1 LYS CA 1 0.25 12.011 ; qtot 0.94
6 opls_140 1 LYS HA 1 0.06 1.008 ; qtot 1
nr: 原子序数type: 原子型resnr: 氨基酸残基数residue: 氨基酸残基名称, 请注意,该残基在 PDB 文件中为“LYS”;使用.rtp条目“LYSH”表示残基被质子化(中性 pH 值的主要状态)。atom: 原子名称cgnr: 电荷组号, 电荷组定义整数电荷的单位;它们有助于加快计算速度charge: 不言自明, “qtot”描述符是分子上电荷的总和mass: 也是不言自明的,typeB, chargeB, massB: 用于自由能微扰(这里不讨论)
后续部分包括[ bonds ], [ pairs ], [ angles ],和 [ dihedrals ]。其中一些部分是不言自明的(键、角和二面角)。GROMACS 手册第 5 章详细介绍了与这些部分相关的参数和函数类型。特殊的 1-4 相互作用包含在“对”下(GROMACS 手册第 5.3.4 节)。
该文件的其余部分涉及定义一些其他有用/必要的拓扑结构,从位置限制开始。“posre.itp”文件由 pdb2gmx 生成;它定义了一个力常数,用于在平衡过程中将原子保持在原位(稍后会详细介绍)。
; Include Position restraint file
#ifdef POSRES
#include "posre.itp"
#endif
这结束了“Protein_A”分子类型的定义。拓扑文件的其余部分专门用于定义其他分子并提供系统级描述。下一个分子类型(默认情况下)是溶剂,在本例中为 SPC/E 水。水的其他典型选择包括 SPC、TIP3P 和 TIP4P。我们通过将“-water spce”传递给 pdb2gmx 来选择它。有关许多不同水模型的精彩总结,请单击此处,但请注意并非所有这些模型都存在于 GROMACS 中。
; Include water topology
#include "oplsaa.ff/spce.itp"
#ifdef POSRES_WATER
; Position restraint for each water oxygen
[ position_restraints ]
; i funct fcx fcy fcz
1 1 1000 1000 1000
#endif
正如你所看到的,水也可以被位置约束,使用力常数( k p r k_{pr} kpr)为1000 kJ m o l − 1 mol^{-1} mol−1 n m − 2 nm_{-2} nm−2。接下来是离子参数
; Include generic topology for ions
#include "oplsaa.ff/ions.itp"
最后是系统级定义。该[ system ]指令给出了将在模拟期间写入输出文件的系统名称。该[ molecules ]指令列出了系统中的所有分子。
[ system ]
; Name
LYSOZYME
[ molecules ]
; Compound #mols
Protein_A
关于该[ molecules ]指令的一些关键说明:
- 所列分子的顺序必须与坐标(在本例中为 .gro)文件中的分子顺序完全匹配。
- 列出的名称必须与[ moleculetype ]每个物种的名称相匹配,而不是残基名称或其他任何名称。
如果您在任何时候未能满足这些具体要求,您将收到来自grompp(稍后讨论)的关于名称不匹配、未找到分子或许多其他错误的致命错误。
现在我们已经检查了拓扑文件的内容,我们可以继续构建我们的系统。
定义晶胞和添加溶剂

现在您已经熟悉了 GROMACS 拓扑的内容,是时候继续构建我们的系统了。在这个例子中,我们将模拟一个简单的水溶液系统。可以在不同溶剂中模拟蛋白质和其他分子,前提是所有涉及的物种都有良好的参数。
定义盒子并用溶剂填充盒子有两个步骤:
- 使用 editconf 模块定义框尺寸。
- 使用溶剂化模块(以前称为 genbox)用水填充盒子。
您现在可以选择如何处理晶胞。出于本教程的目的,我们将使用一个简单的立方体作为单位单元格。随着您对周期性边界条件和盒子类型越来越熟悉,我强烈推荐菱形十二面体,因为它的体积大约是具有相同周期距离的立方盒子的 71%,从而节省了需要添加的水分子数量使蛋白质溶剂化。
让我们使用 editconf 定义框:
gmx editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.0 -bt cubic
上述命令将蛋白质置于方框 (-c) 的中心,并将其放置在距方框边缘至少 1.0 nm 的位置 (-d 1.0)。盒子类型定义为立方体 (-bt cubic)。到盒子边缘的距离是一个重要的参数。由于我们将使用周期性边界条件,因此我们必须满足最小图像约定。也就是说,蛋白质不应该看到它的周期图像,否则计算出的力将是虚假的。指定 1.0 nm 的溶质盒距离意味着蛋白质的任意两个周期图像之间至少有 2.0 nm。这个距离对于模拟中常用的任何cutoff方案都足够了。
现在我们已经定义了一个盒子,我们可以用溶剂(水)填充它。溶剂化是使用溶剂化物完成的:
gmx solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
蛋白质 (-cp) 的配置包含在前面的 editconf 步骤的输出中,溶剂 (-cs) 的配置是标准 GROMACS 安装的一部分。我们使用的是 spc216.gro,这是一个通用的平衡三点溶剂模型。您可以使用 spc216.gro 作为 SPC、SPC/E 或 TIP3P 水的溶剂配置,因为它们都是三点水模型。输出名为 1AKI_solv.gro,我们将拓扑文件 (topol.top) 的名称告诉 solvate,以便对其进行修改。注意[ molecules ]topol.top 指令的变化:
[ molecules ]
; Compound #mols
Protein_A 1
SOL 10832
溶剂化物所做的是跟踪它添加了多少水分子,然后将其写入您的拓扑以反映所做的更改。请注意,如果您使用任何其他(非水)溶剂,溶剂化物不会对您的拓扑结构进行这些更改!它与更新水分子的兼容性是硬编码的。
添加离子

我们现在有一个包含带电蛋白质的溶剂化系统。pdb2gmx 的输出告诉我们该蛋白质的净电荷为 +8 e(基于其氨基酸组成)。如果您在 pdb2gmx 输出中遗漏了此信息,请查看topol.top 中[ atoms ]指令的最后一行;它应该读作(部分)“qtot 8”。由于生命不存在于净电荷下,我们必须向我们的系统中添加离子。
在 GROMACS 中添加离子的工具称为 genion。genion 所做的是通过拓扑读取并用用户指定的离子替换水分子。输入称为运行输入文件,扩展名为.tpr;该文件由 GROMACS grompp 模块(GROMACS处理器)生成,稍后我们运行第一次模拟时也会用到它。grompp 所做的是处理坐标文件和拓扑(描述分子)以生成原子级输入 (.tpr)。.tpr 文件包含系统中所有原子的所有参数。
要使用grompp生成.tpr文件,我们需要一个额外的输入文件,扩展名为 .mdp(分子动态参数文件);grompp 会将 .mdp 文件中指定的参数与坐标和拓扑信息组合起来生成 .tpr 文件。
.mdp 文件通常用于运行能量最小化或 MD 模拟,但在这种情况下仅用于生成系统的原子描述。可以在此处下载示例 .mdp 文件(我们将使用的文件)。
实际上,此步骤中使用的 .mdp 文件可以包含任何合法的参数组合。我通常使用能量最小化脚本,因为它们非常基础,不涉及任何复杂的参数组合。**请注意,本教程提供的文件仅适用于 OPLS-AA 力场。**其他力场的设置,尤其是非键合交互设置会有所不同。
使用以下内容组装 .tpr 文件:
gmx grompp -f ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
现在我们在二进制文件 ions.tpr 中有了我们系统的原子级描述。我们将把这个文件传递给 genion:
gmx genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
出现提示时,选择第 13 组“SOL”来嵌入离子。您不想用离子替换部分蛋白质。
在 genion 命令中,我们提供结构/状态文件作为 (-s) 输入,生成一个 .gro 文件作为(-o)输出 ,处理拓扑 (-p) 以反映水分子的去除和离子的添加,定义正离子和负离子名称(分别为 -pname 和 -nname),并告诉 genion 通过添加正确数量的负离子(-neutral,在这种情况下将添加 8 个 Cl -离子以抵消蛋白质上的 +8 电荷)。除了通过结合指定 -neutral 和 -conc 选项来简单地中和系统之外,您还可以使用 genion 添加指定浓度的离子。有关如何使用这些选项的信息,请参阅 genion 手册页。
使用 -pname 和 -nname 指定的离子名称在 GROMACS 的早期版本中是特定于力场的,但在 4.5 版中已标准化。指定的离子名称始终是所有大写字母的元素符号,这[ moleculetype ]是随后写入拓扑的名称。残基或原子名称可能会或可能不会附加电荷符号 (+/-),具体取决于力场。 不要在 genion 命令中使用原子或残基名称,否则您将在后续步骤中遇到错误。
您的[ molecules ]指令现在应该如下所示:
[ molecules ]
; Compound #mols
Protein_A 1
SOL 10636
CL 8
能量最小化

溶剂化的电子中性体系现在组装好了。在我们开始动态之前,我们必须确保系统没有空间冲突或不适当的几何形状。结构通过能量最小化(EM)过程进行松弛。
EM的过程很像离子的加入。我们将再次使用grompp将结构、拓扑和模拟参数组装到二进制输入文件(.tpr)中,但这一次,我们将通过GROMACS MD引擎mdrun运行能量最小化,而不是将.tpr传递给genion。
使用此输入参数文件,使用grompp组装二进制输入
gmx grompp -f minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
确保您一直在更新您的话题。当运行genbox和genion时,Top文件,否则你会得到很多讨厌的错误消息(“坐标文件中的坐标数不匹配拓扑”等)。
我们现在已经准备好调用mdrun来执行EM
gmx mdrun -v -deffnm em
-v标志是给没有耐心的人用的:它使mdrun变得冗长,这样它就会在每一步都将进程打印到屏幕上。-deffnm标志将定义输入和输出的文件名。因此,如果您没有将grompp输出命名为“em.tpr”,则必须使用mdrun -s标志显式指定其名称。在本例中,我们将获得以下文件:
- em.log:EM进程的ASCII-TEXT日志文件
- em.edr:二进制能源文件
- em.trr: 二进制完整轨迹-
- em.gro:能量最小的结构
要判断EM是否成功,有两个非常重要的因素需要评估。第一个是势能(在EM过程结束时打印,即使没有-v)。Epot应该是负的,并且(对于水中的简单蛋白质)在
1
0
5
−
1
0
6
10^5-10^6
105−106的数量级上,这取决于系统的大小和水分子的数量。第二个重要特征是最大力,
F
m
a
x
F_{max}
Fmax,其目标在mdp中设置为- “emtol = 1000.0”。-表示目标
F
m
a
x
F_{max}
Fmax不大于1000 kJ
m
o
l
−
1
mol^{-1}
mol−1
n
m
−
1
nm^{-1}
nm−1。用
F
m
a
x
>
e
m
t
o
l
F_{max} > emtol
Fmax>emtol可以达到一个合理的Epot。如果发生这种情况,您的系统可能不够稳定,无法进行模拟。评估它可能发生的原因,并可能更改您的最小化参数(integrator,emstep等)。
我们来分析一下。 em.edr 文件包含GROMACS在EM期间收集的所有能量项。您可以使用GROMACS能量模块分析任何.edr文件
gmx energy -f em.edr -o potential.xvg
在提示符下,“10 0”,输入(10)选择电位;输入0终止。你将看到
E
p
o
t
E_pot
Epot的平均值,以及一个名为“potential.xvg”的文件将被写入。要绘制这些数据,您将需要Xmgrace绘图工具。最终的图应该是这样的,展示了
E
p
o
t
E_{pot}
Epot的良好、稳定的收敛

现在我们的系统处于能量最小值,我们可以开始真正的动力学。
平衡

EM 确保我们在几何形状和溶剂方向方面具有合理的起始结构。要开始真正的动力学,我们必须平衡蛋白质周围的溶剂和离子。如果我们在这一点上尝试无限制的动力学,系统可能会崩溃。原因是溶剂主要在自身内部优化,不一定与溶质一起优化。它需要被带到我们希望模拟的温度,并建立关于溶质(蛋白质)的正确方向。在我们达到正确的温度(基于动能)后,我们将向系统施加压力,直到它达到适当的密度。
还记得很久以前 pdb2gmx 生成的 posre.itp 文件吗?我们现在要使用它。posre.itp 的目的是对蛋白质的重原子(任何不是氢的原子)施加位置约束力。允许移动,但前提是要克服大量的能量损失。位置限制的用途是它们允许我们在蛋白质周围平衡我们的溶剂,而不会增加蛋白质结构变化的变量。位置约束的原点(约束势为零的坐标)通过传递给 grompp 的 -r 选项的坐标文件提供。
平衡通常分两个阶段进行。第一阶段在NVT系综下进行(恒定的粒子数、体积和温度)。该系综也称为“等温-等容”或“规范”。这种程序的时间范围取决于系统的内容,但在NVT中,系统的温度应该达到所需值的平台。如果温度尚未稳定,则需要额外的时间。通常,50-100 ps 就足够了,我们将为此练习进行 100-ps NVT平衡。根据您的机器,这可能需要一段时间(如果在 16 个左右的内核上并行运行,则不到一个小时)。在此处获取 .mdp 文件。
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
gmx mdrun -deffnm nvt
除了提供的注释外,还可以在 GROMACS 手册中找到对所用参数的完整解释。记下 .mdp 文件中的一些参数:
gen_vel = yes: 启动速度生成。使用不同的随机种子(gen seed)给出不同的初始速度,因此可以从相同的起始结构进行多个(不同的)模拟。tcoupl = V-rescale: 速度调整温控器是对Berendsen弱耦合方法的改进,Berendsen弱耦合方法不能再现正确的动力学系综。pcoupl = no: 没有施加压力耦合。
让我们分析温度的变化过程,还是用能量
gmx energy -f nvt.edr -o temperature.xvg
在提示下键入“ 16 0”,以选择系统的温度和退出。最终的图应如下所示:

从图中可以清楚地看出,系统的温度迅速达到目标值(300k),并在平衡的剩余部分保持稳定。对于这个系统,一个较短的平衡周期(大约50 ps)可能已经足够了。

上一步,NVT平衡,稳定了系统的温度。在收集数据之前,我们还必须稳定系统的压力(以及密度)。压力平衡是在NPT系综下进行的,其中粒子数、压力和温度都是恒定的。该系综也称为“等温-等压”系综,最接近实验条件。
可在此处找到用于 100-ps NPT平衡的 .mdp 文件。它与用于NVT平衡的参数文件没有太大区别。注意使用 Parrinello-Rahman 恒压器添加压力耦合部分。
其他一些变化:
continuation = yes:我们从NVT平衡阶段 继续模拟gen_vel = no:从轨迹中读取速度(见下文)
gmx grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
gmx mdrun -deffnm npt
让我们再次使用能量来分析压力进程:
gmx energy -f npt.edr -o pressure.xvg
在提示符下输入“18 0”,选择系统压力退出。生成的图形应该如下所示

在 100 ps 平衡阶段,压力值波动很大,但这种行为并不意外。这些数据的运行平均值绘制为图中的红线。在平衡过程中,压力的平均值为 7.5 ± 160.5 巴。请注意,参考压力设置为 1 bar,那么这个结果是否可以接受?压力是一个在 MD 模拟过程中波动很大的量,从大的均方根波动 (160.5 bar) 可以清楚地看出,因此从统计学上讲,人们无法区分所获得的平均值 (7.5 ± 160.5 bar) 之间的差异) 和目标/参考值 (1 bar)。
我们也来看看密度,这次使用能量并在提示符下输入“24 0”。
gmx energy -f npt.edr -o density.xvg

与压力一样,密度的运行平均值也以红色绘制。100 ps 过程中的平均值为 1019 ± 3
k
g
m
−
3
kg m^{ -3}
kgm−3,接近实验值 1000
k
g
m
−
3
kg m^-3
kgm−3和 SPC/E 模型的预期密度 1008
k
g
m
−
3
kg m^-3
kgm−3。SPC/E 水模型的参数与水的实验值非常相似。随着时间的推移,密度值非常稳定,表明系统现在在压力和密度方面达到了很好的平衡。
**请注意:**我经常被问到为什么获得的密度值与我的结果不匹配。与压力相关的条款收敛得很慢,因此你可能不得不运行不扩散核武器条约平衡的时间比这里指定的时间稍长一些。
MD 模拟

完成两个平衡阶段后,系统现在已在所需温度和压力下达到良好平衡。我们现在准备释放位置限制并运行生产 MD 以收集数据。这个过程就像我们之前看到的一样,因为我们将使用检查点文件(在本例中现在包含保留压力耦合信息)到 grompp。我们将运行 1-ns MD 模拟,其脚本可在此处找到。
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr
grompp 将打印 PME 负载的估计值,这将指示应将多少处理器专用于 PME 计算,以及多少处理器用于 PP 计算。有关详细信息,请参阅 GROMACS 4出版物和手册。
Estimate for the relative computational load of the PME mesh part: 0.22
对于立方体盒子,最佳设置的 PME 负载为 0.25(3:1 PP:PME - 我们非常接近最佳!);对于十二面体盒,最佳 PME 负载为 0.33 (2:1 PP:PME)。执行 mdrun 时,程序应自动确定为 PP 和 PME 计算分配的最佳处理器数。因此,请确保为计算指定适当的线程/核心数(-nt X 的值),以便获得最佳性能。
现在,执行 mdrun:
gmx mdrun -deffnm md_0_1
在 GROMACS 2018 中,PME 计算可以卸载到图形处理单元 (GPU),从而大大加快模拟速度。使用 Titan Xp GPU,可以以惊人的 295 ns/天模拟该系统!
在 GPU 上运行 GROMACS
从 4.6 版开始,GROMACS 支持使用 GPU 加速器运行 MD 模拟。随着 2018 版的发布,非键合相互作用和 PME 在 GPU 上计算,仅键合力在 CPU 内核上计算。在构建 GROMACS 时(有关安装说明,请参见www.gromacs.org),如果存在 GPU 硬件,将自动检测到。使用 GPU 加速的最低要求是 CUDA 库和 SDK,以及计算能力 >= 2.0 的 GPU。可以在此处找到一些更常见的 GPU 及其规格的详细列表。
假设您有一个可用的 GPU,使用它的 mdrun 命令非常简单:
gmx mdrun -deffnm md_0_1 -nb gpu
如果您有多个 GPU 可用,或者需要通过 GROMACS 中提供的混合并行化方案自定义工作分配方式,请查阅 GROMACS 手册和网页。此类技术细节超出了本教程的范围。
分析

现在我们已经模拟了我们的蛋白质,我们应该对系统进行一些分析。哪些类型的数据很重要?这是运行模拟之前要问的一个重要问题,因此您应该对要在自己的系统中收集的数据类型有一些想法。对于本教程,将介绍一些基本工具。
第一个是 trjconv,它用作后处理工具,用于去除坐标、校正周期性或手动更改轨迹(时间单位、帧频率等)。对于本练习,我们将使用 trjconv 来解释系统中的任何周期性。蛋白质将扩散通过单元格,并且可能看起来“破碎”或可能“跳”到盒子的另一侧。要说明此类行为,请发出以下命令:
gmx trjconv -s md_0_1.tpr -f md_0_1.xtc -o md_0_1_noPBC.xtc -pbc mol -center
选择 1(“蛋白质”)作为要居中的组,选择 0(“系统”)作为输出。我们将对这条“修正后”的轨迹进行所有分析。我们先来看结构稳定性。GROMACS 有一个称为 rms 的用于 RMSD 计算的内置实用程序。要使用 rms,请发出以下命令:
gmx rms -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rmsd.xvg -tu ns
为最小二乘拟合和用于 RMSD 计算的组选择 4(“主干”)。-tu 标志将以 ns 为单位输出结果,即使轨迹是用 ps 编写的。这样做是为了输出的清晰度(特别是如果你有一个很长的模拟 - 1e+05 ps 看起来不如 100 ns 好)。输出图将显示相对于最小化平衡系统中存在的结构的 RMSD:
如果我们想计算相对于晶体结构的 RMSD,我们可以发出以下命令:
gmx rms -s em.tpr -f md_0_1_noPBC.xtc -o rmsd_xtal.xvg -tu ns
绘制在一起,结果类似于:

两个时间序列均显示 RMSD 水平下降至 ~0.1 nm (1 Å),表明结构非常稳定。图之间的细微差别表明 t = 0 ns 处的结构与该晶体结构略有不同。这是意料之中的,因为它已被能量最小化,并且因为位置限制并非 100% 完美,如前所述。
蛋白质的回转半径是其紧密度的量度。如果蛋白质稳定折叠,它可能会保持相对稳定的Rg值。如果蛋白质展开,其R g将随时间变化。让我们在模拟中分析溶菌酶的回转半径:
gmx gyrate -s md_0_1.tpr -f md_0_1_noPBC.xtc -o gyrate.xvg
选择第 1 组(蛋白质)进行分析。
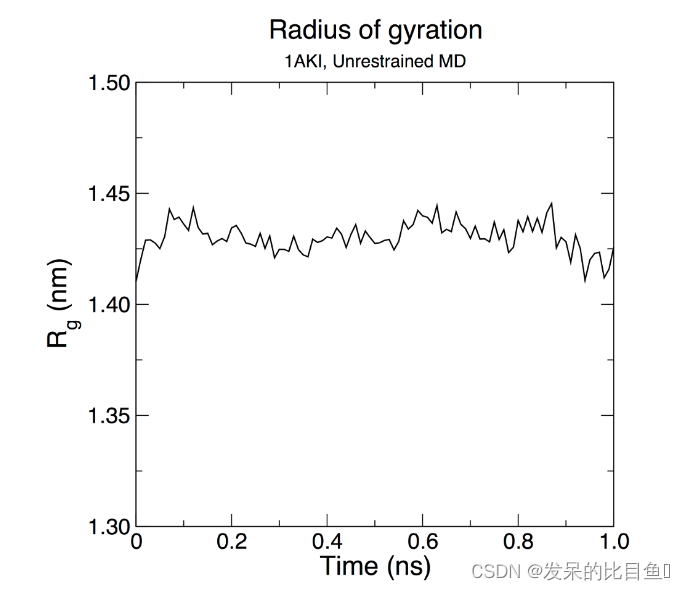
我们可以从合理不变的R g值中看出,蛋白质在 300 K 的 1 ns 过程中以其紧凑(折叠)形式保持非常稳定。这个结果并不意外,但说明了 GROMACS 分析的高级能力内置。