欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://spike.blog.csdn.net/article/details/132410296

AlphaFold2 以其能够以极高的准确度预测蛋白质结构的能力,彻底改变了结构生物学。然而,AlphaFold2 的实现,缺乏训练新模型所需的代码和数据。这些是必要的,用于
- 解决新的任务,如蛋白质-配体复合物结构预测。
- 探索模型学习的过程,这仍然是一个难以理解的问题。
- 评估模型在未见过的折叠空间区域的泛化能力。
OpenFold 是一个快速、节省内存、可训练的 AlphaFold2 的实现,以及 OpenProteinSet 是最大的公开的蛋白质多序列比对数据库。使用 OpenProteinSet 从头开始训练 OpenFold,完全匹配 AlphaFold2 的准确度。在建立了平等性之后,通过使用精心设计的数据集重新训练 OpenFold,评估在折叠空间中泛化的能力。尽管训练集的大小和多样性极度减少,甚至包括几乎完全消除了二级结构元素类别,OpenFold 仍然具有非常强大的泛化能力。通过分析 OpenFold 在训练过程中产生的中间结构,获得了一些令人惊讶的观点,发现模型学习折叠蛋白质的方式,以及发现空间维度是顺序学习的。总而言之,研究展示 OpenFold 的强大和实用性,将成为蛋白质建模社区一个至关重要的新资源。
OpenFold:
- Paper: OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization
- GitHub: https://github.com/aqlaboratory/openfold
- Huggingface: https://huggingface.co/nz/OpenFold
OpenFold 的运行环境配置,参考 开源可训练的蛋白质结构预测框架 OpenFold 的环境配置
使用相同的 MSA 文件,测试 OpenFold 的不同模型与 AlphaFold2 的差异,OpenFold 支持 AlphaFold2 的默认模型与 Finetuning 的模型,比较预测的蛋白质结构之间的差异,由于随机种子与模型架构 (Tensorflow 或 PyTorch) 的差异,实验结果,仅作为参考。
| T1104-A117 | model | TMScore | RMSD | Template |
|---|---|---|---|---|
| alphafold2 | model_1_ptm_pred_0 | 0.8760 | 1.83 | 2022-04-01 |
| unrelaxed_model_1_pred_0 | 0.9036 | 1.66 | 2022-04-01 | |
| openfold | model_1_ptm | 0.5663 | 3.82 | 2022-04-01 |
| model_1 | 0.8625 | 1.91 | 2022-04-01 | |
| model_1_ptm_finetune | 0.9001 | 1.44 | 2022-04-01 | |
| model_1_finetune | 0.8114 | 2.40 | 2022-04-01 |
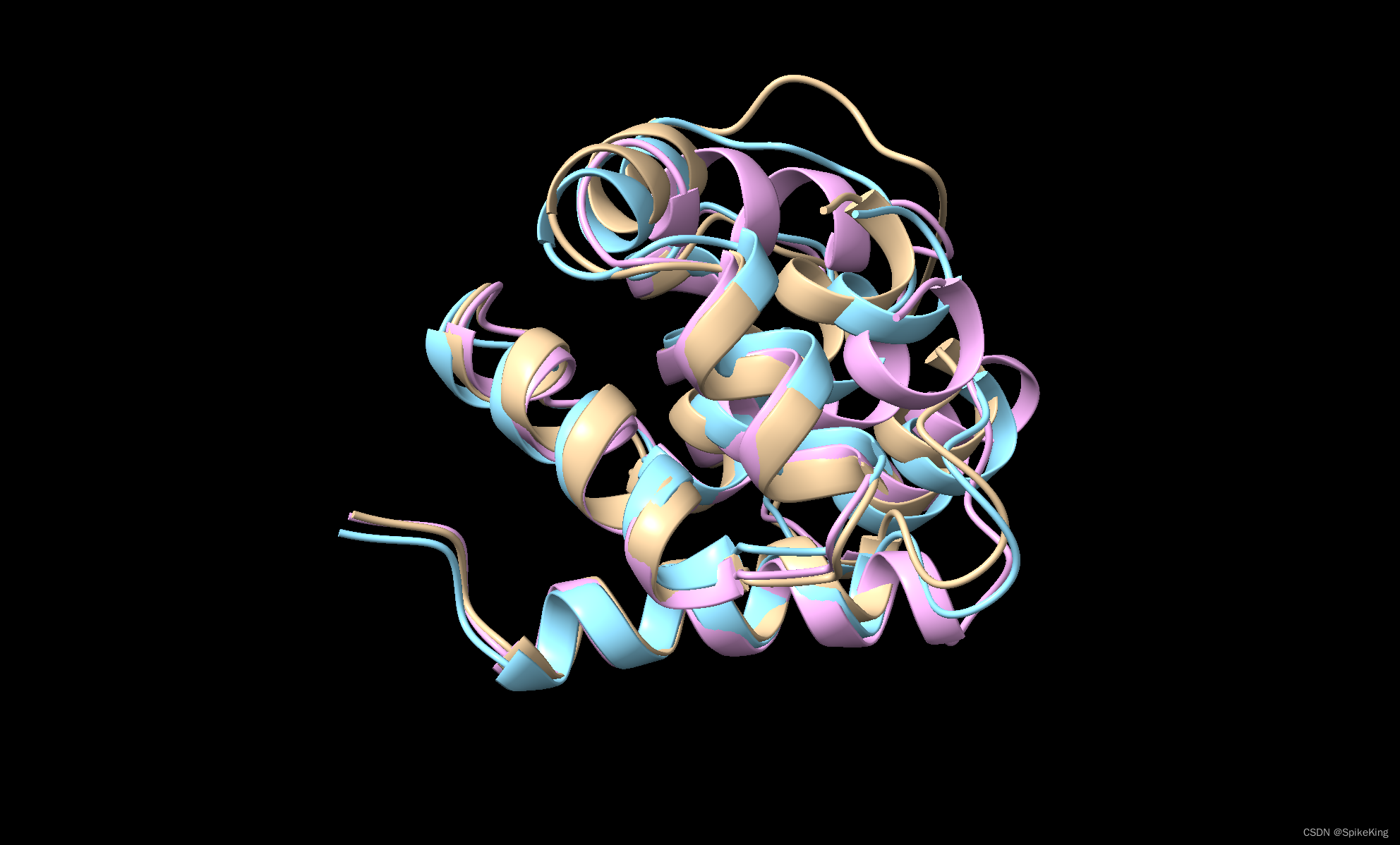
其中,不同结构的比较,黄色是真实结构,蓝色来源于 AlphaFold2 的 monomer_ptm 模型,粉色来源于 OpenFold 的 finetuning_monomer_ptm 模型,即:
1. 模型测试
测试序列 T1104-D1_A117.fasta 来自于 CASP15,即:
>A
QLEDSEVEAVAKGLEEMYANGVTEDNFKNYVKNNFAQQEISSVEEELNVNISDSCVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG
MSA 来自于 AF2 的搜索结果,合计包括 380 条序列,即:
bfd_uniref_hits.a3m
mgnify_hits.sto
pdb_hits.hhr
uniref90_hits.sto
MSA 中 序列数量如下,来自于 AlphaFold2 的日志,即:
Uniref90 MSA size: 29 sequences.
BFD MSA size: 333 sequences.
MGnify MSA size: 28 sequences.
Final (deduplicated) MSA size: 380 sequences.
1.1 AlphaFold2 Monomer pTM 实验
使用 AlphaFold2 的 monomer_ptm 与 monomer 模型,作为对照实验,测试命令:
- FASTA 路径:
mydata/openfold_test/monomer_fasta/T1104-D1_A117.fasta - MSA 与 outputs 路径:
mydata/openfold_test/monomer_ptm_outputs/、mydata/openfold_test/monomer_outputs/,使用已搜索的 MSA 文件夹 - 只使用
pred_0结构,作为对照。
即:
bash run_alphafold.sh -o mydata/openfold_test/monomer_ptm_outputs/ -f mydata/openfold_test/monomer_fasta/T1104-D1_A117.fasta -m monomer_ptm -c full_dbs
bash run_alphafold.sh -o mydata/openfold_test/monomer_ptm_outputs/ -f mydata/openfold_test/monomer_fasta/T1104-D1_A117.fasta -m monomer -c full_dbs
其中,日志如下:
- 模版日期:
2022-04-01 - MSA 数量:
Final (deduplicated) MSA size: 380 sequences.
1.2 OpenFold 基于 AF2 模型参数 实验
测试命令:
- FASTA 路径:
mydata/openfold_test/monomer_fasta/T1104-D1_A117.fasta - MSA 与 outputs 路径:
mydata/openfold_test/monomer_ptm_outputs/、mydata/openfold_test/monomer_outputs/ - 在 OpenFold 中默认的
max_template_date与当前日期相同,因此,需要指定2022-04-01
即:
python3 run_pretrained_openfold.py \
mydata/openfold_test/monomer_fasta/ \
af2-data-v230/pdb_mmcif/mmcif_files \
--uniref90_database_path af2-data-v230/uniref90/uniref90.fasta \
--mgnify_database_path af2-data-v230/mgnify/mgy_clusters_2022_05.fa \
--pdb70_database_path af2-data-v230/pdb70/pdb70 \
--uniclust30_database_path deepmsa2/uniclust30/uniclust30_2018_08 \
--output_dir mydata/openfold_test/monomer_ptm_outputs/T1104-D1_A117/ \
--bfd_database_path af2-data-v230/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt \
--model_device "cuda:0" \
--jackhmmer_binary_path /opt/openfold/hhsuite-speed/jackhmmer \
--hhblits_binary_path /opt/conda/envs/openfold/bin/hhblits \
--hhsearch_binary_path /opt/conda/envs/openfold/bin/hhsearch \
--kalign_binary_path /opt/conda/envs/openfold/bin/kalign \
--config_preset "model_1_ptm" \
--jax_param_path af2-data-v230/params/params_model_1_ptm.npz \
--max_template_date 2022-04-01
测试非 ptm 版本:
#...
--config_preset "model_1" \
--jax_param_path af2-data-v230/params/params_model_1.npz \
#...
1.3 OpenFold 基于 Finetuning 模型参数 实验
模型来源于 Huggingface 地址,即:
finetuning_ptm_2:Checkpoints in chronological order corresponding to peaks in the pTM training phase of the mainline branch. Models in this category include the pTM module and comprise the most recent of the checkpoints in said branch.- 按时间顺序排列的检查点,对应于主线分支的 pTM 训练阶段的峰值。 此类别中的模型,包括 pTM 模块,并且包含所述分支中最新的检查点。
测试命令:
# ...
--config_preset "model_1_ptm" \
--openfold_checkpoint_path openfold/resources/openfold_params/finetuning_ptm_2.pt \
# ...
测试基础版本 model_1 的 finetuning_5.pt 模型,命令如下:
# ...
--config_preset "model_1" \
--openfold_checkpoint_path openfold/resources/openfold_params/finetuning_5.pt \
# ...
2. 源码简读
将参数 openfold_checkpoint_path 修改成 jax_param_path,添加 AF2 的模型地址,即可推理,如下:
af2-data-v230/params/params_model_1_ptm.npz
测试命令:
python3 run_pretrained_openfold.py \
mydata/test \
af2-data-v230/pdb_mmcif/mmcif_files \
--uniref90_database_path af2-data-v230/uniref90/uniref90.fasta \
--mgnify_database_path af2-data-v230/mgnify/mgy_clusters_2022_05.fa \
--pdb70_database_path af2-data-v230/pdb70/pdb70 \
--uniclust30_database_path deepmsa2/uniclust30/uniclust30_2018_08 \
--output_dir mydata/output \
--bfd_database_path af2-data-v230/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt \
--model_device "cuda:0" \
--jackhmmer_binary_path /opt/openfold/hhsuite-speed/jackhmmer \
--hhblits_binary_path /opt/conda/envs/openfold/bin/hhblits \
--hhsearch_binary_path /opt/conda/envs/openfold/bin/hhsearch \
--kalign_binary_path /opt/conda/envs/openfold/bin/kalign \
--config_preset "model_1_ptm" \
--jax_param_path af2-data-v230/params/params_model_1_ptm.npz
输出日志:
INFO:openfold/utils/script_utils.py:Successfully loaded JAX parameters at af2-data-v230/params/params_model_1_ptm.npz...
INFO:run_pretrained_openfold.py:Using precomputed alignments for A at mydata/output/alignments...
INFO:openfold/utils/script_utils.py:Running inference for A...
INFO:openfold/utils/script_utils.py:Inference time: 10.29592083580792
INFO:run_pretrained_openfold.py:Output written to mydata/output/predictions/A_model_1_ptm_unrelaxed.pdb...
INFO:run_pretrained_openfold.py:Running relaxation on mydata/output/predictions/A_model_1_ptm_unrelaxed.pdb...
INFO:openfold/utils/script_utils.py:Relaxation time: 6.760884702205658
INFO:openfold/utils/script_utils.py:Relaxed output written to mydata/output/predictions/A_model_1_ptm_relaxed.pdb...
MSA 部分的结构,outputs/alignments/A,即:
- 没有单独的 fasta 同名文件夹。
- 在 alignments 文件夹中,包括链名 A,之后就是 msa 文件。
- 默认搜索格式都是 msa 格式,而 af2 部分是 sto 格式。
即:
bfd_uniclust_hits.a3m
mgnify_hits.a3m
pdb70_hits.hhr
uniref90_hits.a3m
在源码 run_pretrained_openfold.py 中,搜索 MSA 的部分,如下:
tags是链名,即['A']seqs是序列,即['QLEDS...AVQCG']precompute_alignments是预计算 MSA 文件的方法。
即
# ...
logger.info(f"[CL] tags: {tags}, seqs: {seqs}")
# Does nothing if the alignments have already been computed
precompute_alignments(tags, seqs, alignment_dir, args)
# feature_dicts 第1次是空的,随着循环进行,逐步缓存已有的tag (链名)
feature_dict = feature_dicts.get(tag, None)
if feature_dicts:
logger.info(f"[CL] feature_dict.keys: {feature_dict.keys()}")
# ...
在源码 run_pretrained_openfold.py 中,处理特征的部分,如下:
- 临时 fasta 文件:
process_fasta - tmp_fasta_path: mydata/output/tmp_26020.fasta,其中 26020 是 pid,即进程 id,避免重复。
即:
local_alignment_dir = os.path.join(alignment_dir, tag)
logger.info(f"[CL] process_fasta - tmp_fasta_path: {tmp_fasta_path}")
feature_dict = data_processor.process_fasta(
fasta_path=tmp_fasta_path, alignment_dir=local_alignment_dir
)
在源码 openfold/data/data_pipeline.py 中,process_fasta() 是处理特征的部分,如下:
- 特征包括:
template_features模版特征、sequence_features序列特征、msa_featuresMSA 特征。 template_features模版特征包括:template_aatype、template_all_atom_mask、template_all_atom_positions、template_domain_names、template_sequence、template_sum_probs等 6 个部分。sequence_features序列特征包括:aatype、between_segment_residues、domain_name、residue_index、seq_length、sequence等 6 个部分。msa_featuresMSA 特征包括:deletion_matrix_int、msa、num_alignments等 3 个部分。- 这个特征,是整体特征预处理的核心。
即:
def process_fasta(
self,
fasta_path: str,
alignment_dir: str,
alignment_index: Optional[str] = None,
) -> FeatureDict:
"""Assembles features for a single sequence in a FASTA file"""
with open(fasta_path) as f:
fasta_str = f.read()
input_seqs, input_descs = parsers.parse_fasta(fasta_str)
if len(input_seqs) != 1:
raise ValueError(
f"More than one input sequence found in {fasta_path}."
)
input_sequence = input_seqs[0]
input_description = input_descs[0]
num_res = len(input_sequence)
hits = self._parse_template_hits(alignment_dir, alignment_index)
template_features = make_template_features(
input_sequence,
hits,
self.template_featurizer,
)
logger.info(f"[CL] template_features: {template_features.keys()}")
sequence_features = make_sequence_features(
sequence=input_sequence,
description=input_description,
num_res=num_res,
)
logger.info(f"[CL] sequence_features: {sequence_features.keys()}")
msa_features = self._process_msa_feats(alignment_dir, input_sequence, alignment_index)
logger.info(f"[CL] msa_features: {msa_features.keys()}")
return {
**sequence_features,
**msa_features,
**template_features
}
日志:
run_pretrained_openfold.py:Using precomputed alignments for A at mydata/output/alignments...
run_pretrained_openfold.py:[CL] generate_feature_dict - alignment_dir: mydata/output/alignments
run_pretrained_openfold.py:[CL] len(seqs): 1, seqs: ['QLEDSEVEAVAKGLEEMYANGVTEDNFKNYVKNNFAQQEISSVEEELNVNISDSCVANKIKDEFFAMISISAIVKAAQKKAWKELAVTVLRFAKANGLKTNAIIVAGQLALWAVQCG']
run_pretrained_openfold.py:[CL] process_fasta - tmp_fasta_path: mydata/output/tmp_26020.fasta
openfold/data/data_pipeline.py:[CL] template_features: dict_keys(['template_aatype', 'template_all_atom_mask', 'template_all_atom_positions', 'template_domain_names', 'template_sequence', 'template_sum_probs'])
openfold/data/data_pipeline.py:[CL] sequence_features: dict_keys(['aatype', 'between_segment_residues', 'domain_name', 'residue_index', 'seq_length', 'sequence'])
openfold/data/data_pipeline.py:[CL] msas size: 3
openfold/data/data_pipeline.py:[CL] msa_features['msa']: (260, 117)
openfold/data/data_pipeline.py:[CL] msa_features: dict_keys(['deletion_matrix_int', 'msa', 'num_alignments'])