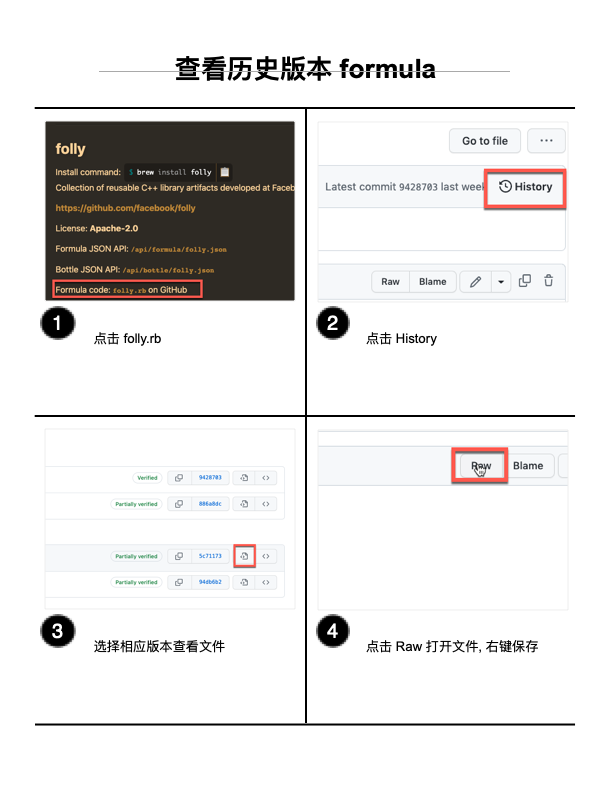
如何利用scGen进行扰动响应预测
scGen是什么
scGen is a generative model to predict single-cell perturbation response across cell types, studies and species (Nature Methods, 2019). scGen is implemented using the scvi-tools framework.
文章传送门
Original tutorial link
相比于原教程,本文主要是对部分步骤的意义进行了注解,方便对流程的理解。
流程
①导包
import logging
import scanpy as sc
import scgen
②准备数据集
train = sc.read("./tests/data/train_kang.h5ad",
backup_url='https://drive.google.com/uc?id=1r87vhoLLq6PXAYdmyyd89zG90eJOFYLk')
train_new = train[~((train.obs["cell_type"] == "CD4T") &
(train.obs["condition"] == "stimulated"))]
数据类型还是Anndata数据。
我们需要制作一个训练数据集和一个测试数据集,并且需要给数据标注上condition和cell_type。
其中condition只能有两种标签,分别代表响应状态和未响应状态,如’control’和’stimulated’。
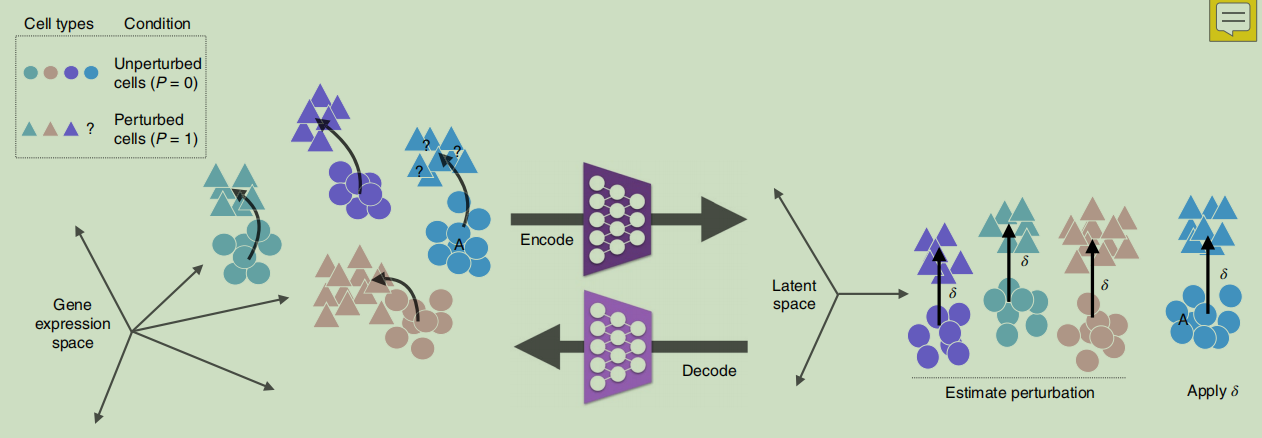
scGen就是用在latent space中学到的从control到stimulated的映射向量来进行perturbation response prediction的。
映射结果是RNA expression matrix。
如原理下图:
③模型训练
scgen.SCGEN.setup_anndata(train_new, batch_key="condition", labels_key="cell_type")
model = scgen.SCGEN(train_new)
model.train(
max_epochs=100,
batch_size=32,
early_stopping=True,
early_stopping_patience=25
)
# Save the trained model
model.save("../saved_models/model_perturbation_prediction.pt", overwrite=True)
查看latent space的数据分布
latent_X = model.get_latent_representation()
latent_adata = sc.AnnData(X=latent_X, obs=train_new.obs.copy())
sc.pp.neighbors(latent_adata)
sc.tl.umap(latent_adata)
sc.pl.umap(latent_adata, color=['condition', 'cell_type'], wspace=0.4, frameon=False)
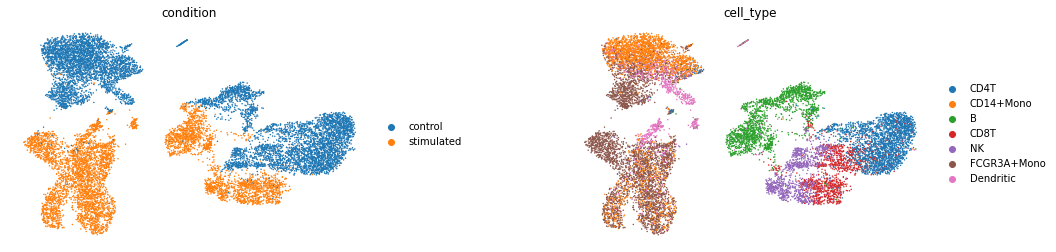
理想的情况应该是模型在隐层空间中对condition做出了较好的区分,这样模型才能有效的学到从control到stimulated的映射向量
④预测
pred, delta = model.predict(
ctrl_key='control',
stim_key='stimulated',
celltype_to_predict='CD4T'
)
pred.obs['condition'] = 'pred'
⑤验证预测结果
PCA
ctrl_adata = train[((train.obs['cell_type'] == 'CD4T') & (train.obs['condition'] == 'control'))]
stim_adata = train[((train.obs['cell_type'] == 'CD4T') & (train.obs['condition'] == 'stimulated'))]
eval_adata = ctrl_adata.concatenate(stim_adata, pred)
sc.tl.pca(eval_adata)
sc.pl.pca(eval_adata, color="condition", frameon=False)
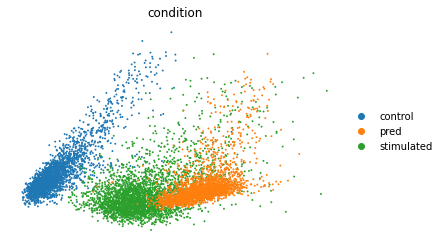
这里是通过绘制RNA表达矩阵PCA降维后的散点图来进行验证,由于我们的预测结果是stimulated,理想的情况下预测结果应该是靠近stimulated并远离control。
Mean correlation plot
Plots mean matching figure for a set of specific genes.
CD4T = train[train.obs["cell_type"] =="CD4T"]
sc.tl.rank_genes_groups(CD4T, groupby="condition", method="wilcoxon")
diff_genes = CD4T.uns["rank_genes_groups"]["names"]["stimulated"]
print(diff_genes)
r2_value = model.reg_mean_plot(
eval_adata,
axis_keys={"x": "pred", "y": "stimulated"},
# list of gene names to be plotted.
gene_list=diff_genes[:10],
labels={"x": "predicted", "y": "ground truth"},
path_to_save="./reg_mean1.pdf",
show=True,
legend=False
)
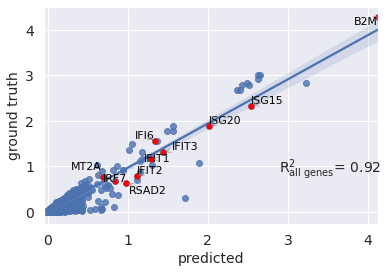
这里是用训练数据中的基因平均表达量作y,预测数据的基因平均表达量作x,作线性回归并计算决定系数 R 2 R^2 R2, R 2 R^2 R2值越靠近1表示基因表达越相近。(这里是计算了全部基因的决定系数)
r2_value = model.reg_mean_plot(
eval_adata,
axis_keys={"x": "pred", "y": "stimulated"},
gene_list=diff_genes[:10],
top_100_genes= diff_genes,
labels={"x": "predicted","y": "ground truth"},
path_to_save="./reg_mean1.pdf",
show=True,
legend=False
)
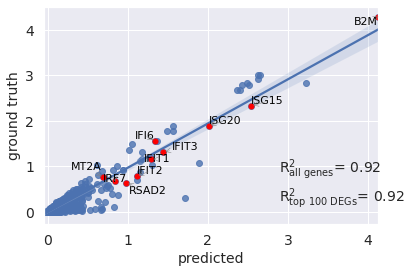
和上图不同,这里还额外计算了condition差异表达基因的决定系数。
Violin plot for a specific gene
sc.pl.violin(eval_adata, keys="ISG15", groupby="condition")
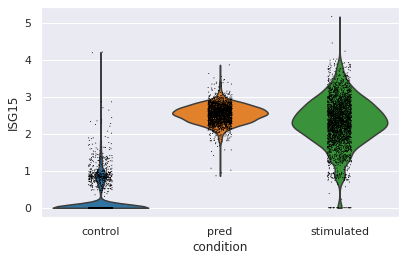
这里绘制了重点关注基因ISG15的琴谱图,可以看出预测结果的细胞分布和stimulated相似。