R语言在RNA-seq中的应用
文章目录
- R语言在RNA-seq中的应用
- 生成工作流环境
- 读取和处理数据
- 由targets文件提供实验定义
- 对实验数据进行质量过滤和修剪
- 生成FASTQ质量报告
- 比对
- 建立HISAT2索引并比对
- 读长量化
- 读段计数
- 样本间的相关性分析
- 差异表达分析
- 运行edgeR
- 可视化差异表达结果
- 计算和绘制差异表达基因(DEG)集合的Venn图
- GO富集分析
- 准备工作
- 批量进行GO富集分析
- 绘制批量GO富集分析的结果
- 层次聚类和热图绘制
生成工作流环境
之后代码运行可能会有网络问题,通过set_config函数设置即可。
.libPaths("D:/000大三下/R语言/实验/Lab4/lab4packages")
library(httr)
set_config(
use_proxy(url="127.0.0.1", port=7890)
)
library(systemPipeR)
library(systemPipeRdata)
#####################################################
## 1. Generate workflow environment
#####################################################
setwd(choose.dir())
genWorkenvir(workflow = "rnaseq")
setwd("rnaseq")
读取和处理数据
由targets文件提供实验定义
读取和预处理实验数据。具体步骤如下:
- 首先,使用
system.file函数找到targets.txt文件的路径,这个文件包含了所有的FASTQ文件和样本比较的信息。 - 然后,使用
read.delim函数读取targets.txt文件,忽略以#开头的注释行,并只保留前四列。 - 最后,打印出
targets对象,查看数据的结构和内容。
## 2. Read preprocessing
#####################################################
## 2.1 Experiment definition provided by targets file
## The targets file defines all FASTQ files and sample comparisons
## of the analysis workflow.
targetspath <- system.file("extdata", "targets.txt", package = "systemPipeR")
targets <- read.delim(targetspath, comment.char = "#")[, 1:4]
targets
对实验数据进行质量过滤和修剪
对实验数据进行质量过滤和修剪。具体步骤如下:
- 首先,使用
loadWorkflow函数从cwl和yml参数文件以及targets文件中构建一个SYSargs2对象,这个对象包含了执行trimLRPatterns函数的所有参数和输入输出路径。 - 然后,使用
renderWF函数根据targets文件中的文件名和样本名替换cwl和yml文件中的占位符,生成一个完整的工作流对象。 - 接着,打印出
trim对象,查看工作流的各个组成部分。 - 最后,使用
output函数查看输出路径中的前两个修剪后的FASTQ文件。
## 2.2 Read quality filtering and trimming
## The function preprocessReads allows to apply predefined or custom read
## preprocessing functions to all FASTQ files referenced in a SYSargs2 container, such
## as quality filtering or adapter trimming routines. The paths to the resulting output
## FASTQ files are stored in the output slot of the SYSargs2 object. The following
## example performs adapter trimming with the trimLRPatterns function from the
## Biostrings package. After the trimming step a new targets file is generated (here
## targets_trim.txt) containing the paths to the trimmed FASTQ files. The new
## targets file can be used for the next workflow step with an updated SYSargs2
## instance, e.g. running the NGS alignments using the trimmed FASTQ files.
## First,we construct SYSargs2 object from cwl and yml param and targets files.
dir_path <- system.file("extdata/cwl/preprocessReads/trim-se",
package = "systemPipeR")
trim <- loadWorkflow(targets = targetspath, wf_file = "trim-se.cwl",
input_file = "trim-se.yml", dir_path = dir_path)
trim <- renderWF(trim, inputvars = c(FileName = "_FASTQ_PATH1_",
SampleName = "_SampleName_"))
trim
output(trim)[1:2]
生成FASTQ质量报告
生成FASTQ质量报告。具体步骤如下:
- 首先,使用
seeFastq函数对trim对象中的输入文件进行质量分析,计算每个文件的碱基质量分布、序列长度分布、GC含量分布和k-mer频率分布。 - 然后,使用
pdf函数创建一个PDF文件,用于保存质量报告的图形。 - 接着,使用
seeFastqPlot函数绘制质量报告的图形,包括每个文件的四个子图。 - 最后,使用
dev.off函数关闭PDF设备,完成图形的保存。
## 2.3 FASTQ quality report
fqlist <- seeFastq(fastq = infile1(trim), batchsize = 10000,
klength = 8)
pdf("./results/fastqReport.pdf", height = 18, width = 4 * length(fqlist))
seeFastqPlot(fqlist)
dev.off()
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-8o8Xh8q9-1685677405310)(D:/000%E5%A4%A7%E4%B8%89%E4%B8%8B/R%E8%AF%AD%E8%A8%80/%E5%AE%9E%E9%AA%8C/Lab4/rnaseq/results/fastqReport.png)]
比对
建立HISAT2索引并比对
使用HISAT2进行短读比对的步骤。主要包括以下几个步骤:
- 构建HISAT2索引:首先,代码中使用
loadWorkflow函数加载了一个工作流程对象(idx),并指定了索引构建的相关参数。然后,通过调用renderWF函数渲染工作流程对象,并通过cmdlist函数获取相应的命令列表。最后,使用runCommandline函数运行命令列表来构建HISAT2索引。 - 进行映射:接下来,代码中使用
loadWorkflow函数加载了另一个工作流程对象(args),并指定了映射的相关参数。通过调用renderWF函数渲染工作流程对象,将文件路径和样本名称作为输入变量进行替换。然后,通过调用cmdlist函数获取命令列表,以及通过output函数获取输出结果的相关信息。 - 运行命令行模式:在代码中使用
runCommandline函数来运行命令列表,以进行短读比对。 - 处理输出文件:在代码中使用
output_update函数来修改args对象,以模拟对生成的对齐结果文件进行处理。具体操作是将输出文件的后缀名修改为".sam"和".bam",并将文件路径中的目录设置为FALSE,以方便后续处理。 - 检查生成的BAM文件:最后,代码中使用
subsetWF函数选择输出结果中的BAM文件路径,并通过file.exists函数检查这些文件是否存在。
## 3.1 Read mapping with HISAT2
## The following steps will demonstrate how to use the short read aligner Hisat2 (Kim,
## Langmead, and Salzberg 2015) in both interactive job submissions and batch
## submissions to queuing systems of clusters using the systemPipeR's new CWL
## command-line interface.
## First, build HISAT2 index. (Skip this step)
#dir_path <- system.file("extdata/cwl/hisat2/hisat2-idx", package = "systemPipeR")
#idx <- loadWorkflow(targets = NULL, wf_file = "hisat2-index.cwl",
# input_file = "hisat2-index.yml", dir_path = dir_path)
#idx <- renderWF(idx)
#idx
#cmdlist(idx)
#runCommandline(idx, make_bam = FALSE)
## Second, mapping.
dir_path <- system.file("extdata/cwl/hisat2", package = "systemPipeR")
args <- loadWorkflow(targets = targetspath, wf_file = "hisat2-mapping-se.cwl",
input_file = "hisat2-mapping-se.yml", dir_path = dir_path)
args <- renderWF(args, inputvars = c(FileName = "_FASTQ_PATH1_",
SampleName = "_SampleName_"))
args
cmdlist(args)[1:2]
output(args)[1:2]
## Runc single Machine
#args <- runCommandline(args) (Skip this)
# Move all *bam files from Lab_4 files/Bam to rnaseq/results
# This command is used to modify class "args" to simulate alignment. (Weihan)
args <- output_update(args, dir = FALSE, replace = TRUE,
extension = c(".sam", ".bam"))
## Check whether all BAM files have been created.
outpaths <- subsetWF(args, slot = "output", subset = 1, index = 1)
file.exists(outpaths)
读长量化
读段计数
使用多核心并行模式下的summarizeOverlaps函数进行读段计数的过程。主要包括以下几个步骤:
-
导入所需的包:代码中使用
library函数导入了GenomicFeatures和BiocParallel包,用于进行基因组特征和并行计算的操作。 -
创建转录组数据库:通过
makeTxDbFromGFF函数,根据GFF文件创建了一个转录组数据库对象(txdb),用于存储基因组注释信息。 -
读取比对结果:通过
readGAlignments函数读取比对结果文件(BAM文件)并存储到变量align中,展示了如何将BAM文件读取到R中进行后续处理。 -
定义感兴趣的外显子基因区域:通过
exonsBy函数根据转录组数据库和给定的外显子区域,生成了一个外显子基因区域对象(eByg)。 -
创建BAM文件列表:通过
BamFileList函数创建了一个BAM文件列表对象(bfl),用于存储需要进行读取计数的BAM文件路径。 -
并行计算设置:通过
MulticoreParam函数创建了一个多核心并行计算参数对象(multicoreParam),并通过register函数注册该参数。 -
执行读段计数:通过
bplapply函数以并行计算的方式对BAM文件列表中的每个文件执行summarizeOverlaps函数进行读取计数。计数结果存储在counteByg列表中。 -
处理计数结果:将计数结果转化为数据框形式(
countDFeByg),并设置行名和列名。 -
计算RPKM:通过
apply函数以每列为单位,调用returnRPKM函数计算RPKM值(rpkmDFeByg)。 -
输出结果:使用
write.table函数将计数结果和RPKM结果分别写入到"results/countDFeByg.xls"和"results/rpkmDFeByg.xls"文件中。 -
数据示例展示:使用
read.delim函数读取计数结果和RPKM结果文件的部分数据进行展示。 -
注意:说明了在大多数统计差异表达或丰度分析方法(如edgeR或DESeq2)中,应使用原始计数值作为输入。RPKM值的使用应限制在一些特殊应用中,例如手动比较不同基因或特征的表达水平。
## 4.1 Read counting with summarizeOverlaps in parallel mode using multiple cores
## Reads overlapping with annotation ranges of interest are counted for each sample
## using the summarizeOverlaps function (Lawrence et al. 2013). The read counting is
## preformed for exonic gene regions in a non-strand-specific manner while ignoring
## overlaps among different genes. Subsequently, the expression count values are
## normalized by reads per kp per million mapped reads (RPKM). The raw read count
## table (countDFeByg.xls) and the corresponding RPKM table (rpkmDFeByg.xls) are
## written to separate files in the directory of this project. Parallelization is achieved
## with the BiocParallel package, here using 8 CPU cores.
library("GenomicFeatures")
library(BiocParallel)
txdb <- makeTxDbFromGFF(file = "data/tair10.gff", format = "gff",
dataSource = "TAIR", organism = "Arabidopsis thaliana")
saveDb(txdb, file = "./data/tair10.sqlite")
txdb <- loadDb("./data/tair10.sqlite")
outpaths <- subsetWF(args, slot = "output", subset = 1, index = 1)
(align <- readGAlignments(outpaths[1])) # 报错 Demonstrates how to read bam file into R
eByg <- exonsBy(txdb, by = c("gene"))
bfl <- BamFileList(outpaths, yieldSize = 50000, index = character())
multicoreParam <- MulticoreParam(workers = 2) # Not supported on Windows, don't worry.
register(multicoreParam)
registered()
counteByg <- bplapply(bfl, function(x) summarizeOverlaps(eByg, x, mode = "Union", ignore.strand = TRUE, inter.feature = FALSE, singleEnd = TRUE))
countDFeByg <- sapply(seq(along = counteByg), function(x) assays(counteByg[[x]])$counts)
rownames(countDFeByg) <- names(rowRanges(counteByg[[1]]))
colnames(countDFeByg) <- names(bfl)
rpkmDFeByg <- apply(countDFeByg, 2, function(x) returnRPKM(counts = x,
ranges = eByg))
write.table(countDFeByg, "results/countDFeByg.xls", col.names = NA,
quote = FALSE, sep = "\t")
write.table(rpkmDFeByg, "results/rpkmDFeByg.xls", col.names = NA,
quote = FALSE, sep = "\t")
## Sample of data slice of count table
read.delim("results/countDFeByg.xls", row.names = 1, check.names = FALSE)[1:4,1:5]
## Sample of data slice of RPKM table
read.delim("results/rpkmDFeByg.xls", row.names = 1, check.names = FALSE)[1:4,1:4]
## Note, for most statistical differential expression or abundance analysis methods,
## such as edgeR or DESeq2, the raw count values should be used as input. The usage
## of RPKM values should be restricted to specialty applications required by some
## users, e.g. manually comparing the expression levels among different genes or
## features.
样本间的相关性分析
进行样本间的相关性分析。利用DESeq2包进行样本间相关性分析的过程。首先将计数数据导入,然后构建DESeq2数据集,并计算样本间的Spearman相关系数。最后,通过层次聚类和绘图,将相关性结果以聚类图的形式保存在PDF文件中。主要包括以下几个步骤:
-
导入所需的包:通过
library函数导入了DESeq2和ape包,用于进行差异表达分析和绘制聚类图的操作。 -
读取计数数据:通过
read.table函数将计数结果文件"results/countDFeByg.xls"读取为一个矩阵(countDF)。 -
构建DESeq2数据集:通过
DESeqDataSetFromMatrix函数将计数数据(countDF)和条件数据(colData)构建为一个DESeq2数据集(dds),指定了条件设计。 -
计算相关系数:通过
cor函数计算基于rlog转换后的表达值的Spearman相关系数。将rlog函数应用于DESeq2数据集(dds)的表达值,然后通过assay函数提取表达矩阵,并计算相关系数(d)。 -
层次聚类:通过
hclust函数对距离矩阵(dist(1 - d))进行层次聚类,生成聚类树对象(hc)。 -
绘制聚类图:通过
pdf函数创建一个PDF文件(“results/sample_tree.pdf”),然后使用plot.phylo函数绘制聚类树(as.phylo(hc)),并设置图形的样式参数。 -
保存聚类图:通过
dev.off函数关闭PDF文件,保存并生成聚类图文件。
## 4.2 Sample-wise correlation analysis
## The following computes the sample-wise Spearman correlation coefficients from the
## rlog transformed expression values generated with the DESeq2 package. After
## transformation to a distance matrix, hierarchical clustering is performed with the
## hclust function and the result is plotted as a dendrogram (also see file
## sample_tree.pdf).
library(DESeq2, quietly = TRUE)
library(ape, warn.conflicts = FALSE)
countDF <- as.matrix(read.table("./results/countDFeByg.xls"))
colData <- data.frame(row.names = targets.as.df(targets(args))$SampleName,
condition = targets.as.df(targets(args))$Factor)
dds <- DESeqDataSetFromMatrix(countData = countDF, colData = colData,
design = ~condition)
d <- cor(assay(rlog(dds)), method = "spearman")
hc <- hclust(dist(1 - d))
pdf("results/sample_tree.pdf")
plot.phylo(as.phylo(hc), type = "p", edge.col = "blue", edge.width = 2,
show.node.label = TRUE, no.margin = TRUE)
dev.off()

差异表达分析
运行edgeR
使用edgeR包进行差异表达分析的过程。通过读取计数数据和目标样本信息,定义比较组,运行edgeR分析,并将结果输出到文件中。另外,还使用biomaRt包获取基因描述信息,并将其添加到差异表达结果中。主要包括以下几个步骤:
- 导入所需的包:通过
library函数导入了edgeR和biomaRt包,用于差异表达分析和获取基因描述信息的操作。 - 读取计数数据和目标样本信息:通过
read.delim函数分别读取计数数据文件"results/countDFeByg.xls"和目标样本信息文件"targets.txt"。 - 定义比较组:通过
readComp函数从目标样本信息中提取比较组信息,将其存储在变量cmp中。 - 运行edgeR:通过
run_edgeR函数利用glm方法对计数数据进行差异表达分析。传入计数数据(countDF)、目标样本信息(targets)和比较组信息(cmp[[1]]),并指定independent = FALSE表示非独立比较。 - 添加基因描述信息:通过使用
biomaRt包中的useMart和getBM函数,连接到植物数据库(plants_mart)并获取基因描述信息。将基因描述信息添加到差异表达结果(edgeDF)的数据框中。 - 输出结果:使用
write.table函数将差异表达结果(edgeDF)写入到"./results/edgeRglm_allcomp.xls"文件中,以制表符分隔,不加引号,列名不写入文件。
## 5. Analysis of DEGs
#####################################################
## The analysis of differentially expressed genes (DEGs) is performed with the glm
## method of the edgeR package (Robinson, McCarthy, and Smyth 2010). The sample
## comparisons used by this analysis are defined in the header lines of the
## targets.txt file starting with <CMP>.
## 5.1 Run edgeR
library(edgeR)
countDF <- read.delim("results/countDFeByg.xls", row.names = 1,
check.names = FALSE)
targets <- read.delim("targets.txt", comment = "#")
cmp <- readComp(file = "targets.txt", format = "matrix", delim = "-")
edgeDF <- run_edgeR(countDF = countDF, targets = targets, cmp = cmp[[1]],
independent = FALSE, mdsplot = "")
## Add gene descriptions
library("biomaRt")
m <- useMart("plants_mart", dataset = "athaliana_eg_gene", host = "https://plants.ensembl.org")
desc <- getBM(attributes = c("tair_locus", "description"), mart = m)
desc <- desc[!duplicated(desc[, 1]), ]
descv <- as.character(desc[, 2])
names(descv) <- as.character(desc[, 1])
edgeDF <- data.frame(edgeDF, Desc = descv[rownames(edgeDF)],
check.names = FALSE)
write.table(edgeDF, "./results/edgeRglm_allcomp.xls", quote = FALSE,
sep = "\t", col.names = NA)
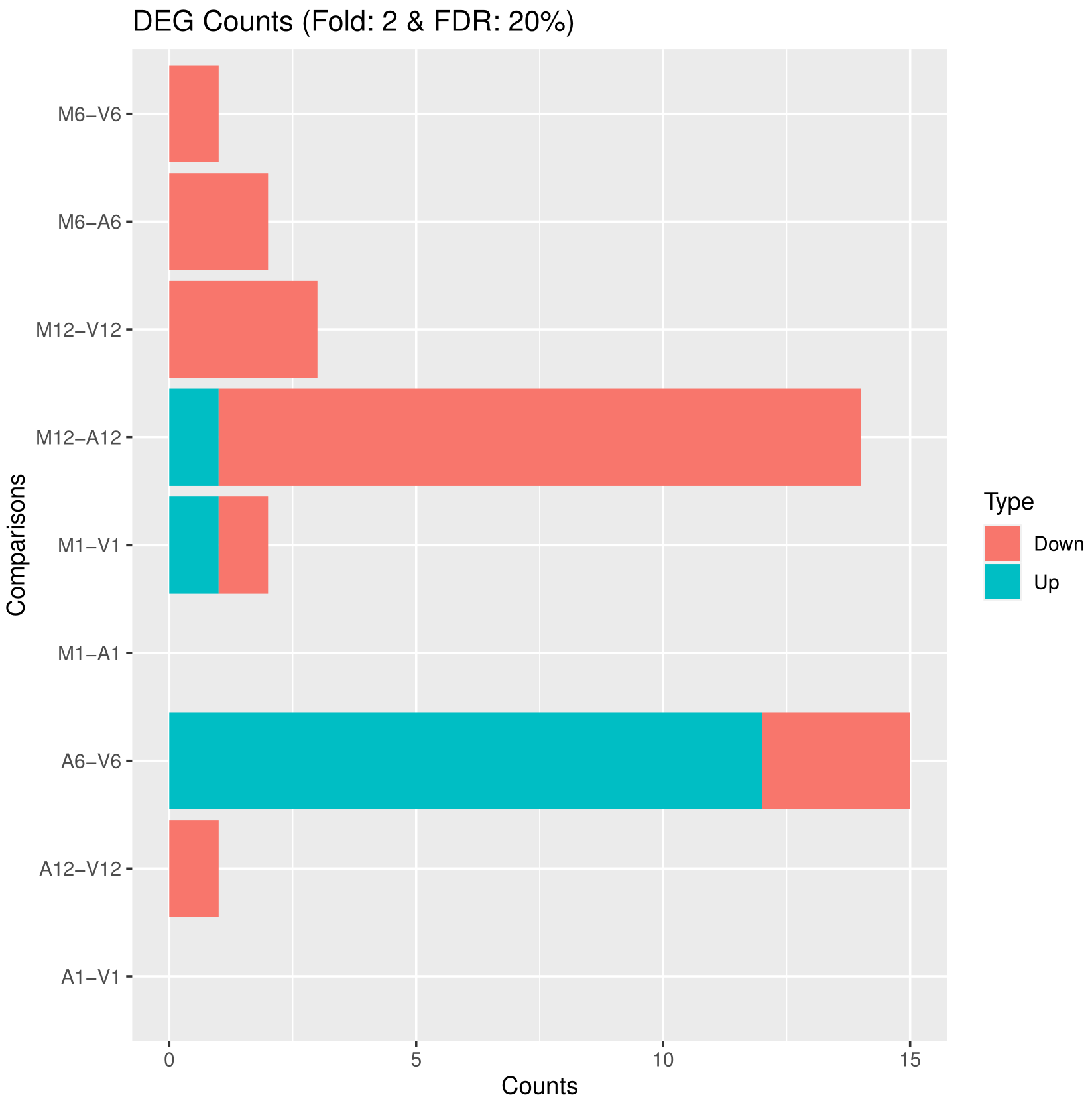
可视化差异表达结果
筛选和可视化差异表达结果,首先读取差异表达结果文件,然后根据设定的筛选条件进行筛选,并将筛选结果绘制成图形保存在PDF文件中。此外,还将筛选后的DEG统计结果输出到文件中。主要包括以下几个步骤:
-
读取差异表达结果:通过
read.delim函数读取差异表达结果文件"results/edgeRglm_allcomp.xls"。 -
绘制DEG结果图:通过
pdf函数创建一个PDF文件(“results/DEGcounts.pdf”),然后使用filterDEGs函数对差异表达结果进行筛选和绘图。筛选条件通过filter参数指定,其中包括折叠变化(Fold)和调整的p值(FDR)。绘图结果保存在PDF文件中。 -
输出DEG统计结果:使用
write.table函数将DEG结果的摘要信息(DEG_list$Summary)写入到"./results/DEGcounts.xls"文件中,以制表符分隔,不加引号,不写入行名。
## 5.2 Plot DEG results
## Filter and plot DEG results for up and down regulated genes. The definition of up
## and down is given in the corresponding help file. To open it, type ?filterDEGs in
## the R console.
edgeDF <- read.delim("results/edgeRglm_allcomp.xls", row.names = 1,
check.names = FALSE)
pdf("results/DEGcounts.pdf")
DEG_list <- filterDEGs(degDF = edgeDF, filter = c(Fold = 2, FDR = 20))
dev.off()
write.table(DEG_list$Summary, "./results/DEGcounts.xls", quote = FALSE,
sep = "\t", row.names = FALSE)
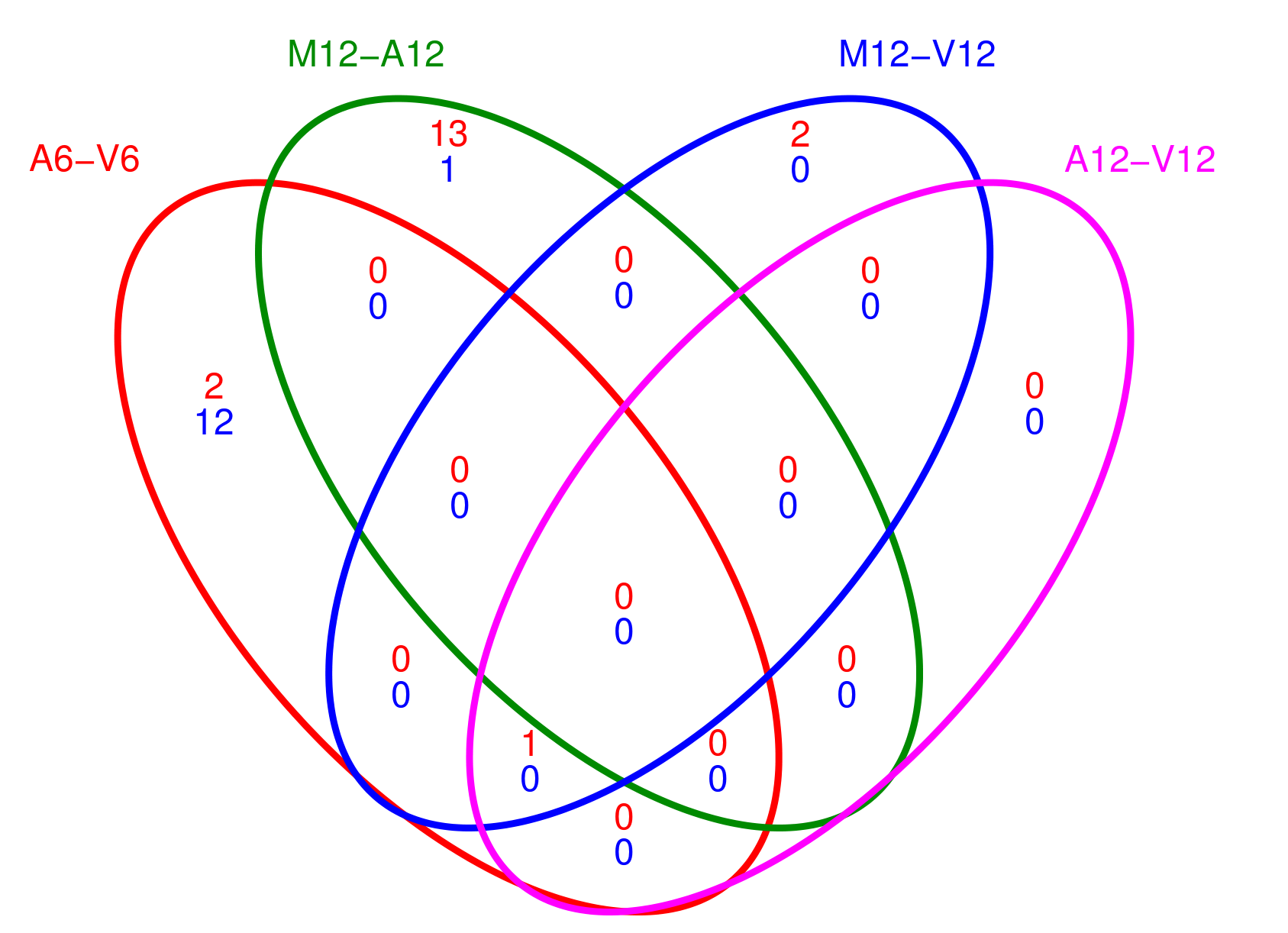
计算和绘制差异表达基因(DEG)集合的Venn图
利用overLapper和vennPlot函数计算和绘制差异表达基因集合的Venn图。首先计算上调和下调基因集合的Venn图交集,并将结果保存。然后根据设定绘制Venn图,并将图形保存到PDF文件中。主要包括以下几个步骤:
-
创建Venn图数据:通过
overLapper函数计算DEG集合的Venn图交集。首先使用DEG_list$Up[6:9]作为输入,表示选取DEG结果中上调基因的第6至第9个集合,然后将结果保存在vennsetup变量中。接着,使用DEG_list$Down[6:9]作为输入,表示选取DEG结果中下调基因的第6至第9个集合,将结果保存在vennsetdown变量中。 -
绘制Venn图:通过
pdf函数创建一个PDF文件(“results/vennplot.pdf”),然后使用vennPlot函数绘制Venn图。将需要绘制的Venn图数据传入list函数中,并通过mymain和mysub参数指定主标题和副标题为空字符串。此外,colmode参数设为2表示使用两种颜色(蓝色和红色)来区分上调和下调的基因集合。 -
结束绘图:通过
dev.off函数结束图形设备,保存Venn图到PDF文件中。
## 5.3 Venn diagrams of DEG sets
## The overLapper function can compute Venn intersects for large numbers of sample
## sets (up to 20 or more) and plots 2-5 way Venn diagrams. A useful feature is the
## possibility to combine the counts from several Venn comparisons with the same
## number of sample sets in a single Venn diagram (here for 4 up and down DEG sets).
vennsetup <- overLapper(DEG_list$Up[6:9], type = "vennsets")
vennsetdown <- overLapper(DEG_list$Down[6:9], type = "vennsets")
pdf("results/vennplot.pdf")
vennPlot(list(vennsetup, vennsetdown), mymain = "", mysub = "",
colmode = 2, ccol = c("blue", "red"))
dev.off()
GO富集分析
准备工作
进行基因-基因本体(Gene Ontology, GO)富集分析的准备工作。首先选择并获取BioMart数据库,然后从数据库中获取基因到GO的映射关系,并对结果进行预处理。最后,创建基因到GO的CATdb对象用于后续的GO富集分析。主要包括以下几个步骤:
-
选择和获取BioMart数据库:通过使用
listMarts函数列出可用的BioMart数据库,选择目标数据库。在这里,通过指定host参数为"https://plants.ensembl.org"来获取与植物相关的数据库。然后使用useMart函数选择目标数据库和数据集。 -
获取基因到GO映射:通过使用
getBM函数从选择的BioMart数据库中获取基因到GO的映射关系。指定所需的属性(attributes)为"go_id"(GO标识符)、“tair_locus”(基因标识符)和"namespace_1003"(GO的命名空间)。将获取的结果保存在go变量中。 -
数据预处理:对获取的基因到GO映射进行预处理。首先,去除命名空间为空的条目。然后,将命名空间的值转换为缩写形式("F"表示分子功能,"P"表示生物过程,"C"表示细胞组分)。最后,将结果保存在文件"GOannotationsBiomart_mod.txt"中。
-
创建基因到GO的CATdb对象:通过使用
makeCATdb函数创建基因到GO的CATdb对象。指定文件路径、列号以及其他必要的参数。将创建的CATdb对象保存在文件"catdb.RData"中。
## 6. GO term enrichment analysis
#####################################################
## 6.1 Obtain gene-to-GO mappings
## The following shows how to obtain gene-to-GO mappings from biomaRt (here for
## A. thaliana) and how to organize them for the downstream GO term enrichment
## analysis. Alternatively, the gene-to-GO mappings can be obtained for many
## organisms from Bioconductor’s *.db genome annotation packages or GO
## annotation files provided by various genome databases. For each annotation this
## relatively slow preprocessing step needs to be performed only once. Subsequently,
## the preprocessed data can be loaded with the load function as shown in the next
## subsection.
library("biomaRt")
listMarts() # To choose BioMart database
listMarts(host = "https://plants.ensembl.org")
m <- useMart("plants_mart", host = "https://plants.ensembl.org")
listDatasets(m)
m <- useMart("plants_mart", dataset = "athaliana_eg_gene", host = "https://plants.ensembl.org")
listAttributes(m) # Choose data types you want to download
go <- getBM(attributes = c("go_id", "tair_locus", "namespace_1003"),
mart = m)
# If download fail, you can load the following Rdata.
#load("Lab_4 files/go.Rdata")
go <- go[go[, 3] != "", ]
go[, 3] <- as.character(go[, 3])
go[go[, 3] == "molecular_function", 3] <- "F"
go[go[, 3] == "biological_process", 3] <- "P"
go[go[, 3] == "cellular_component", 3] <- "C"
go[1:4, ]
# dir.create("./data/GO")
write.table(go, "data/GO/GOannotationsBiomart_mod.txt", quote = FALSE,
row.names = FALSE, col.names = FALSE, sep = "\t")
catdb <- makeCATdb(myfile = "data/GO/GOannotationsBiomart_mod.txt",
lib = NULL, org = "", colno = c(1, 2, 3), idconv = NULL)
save(catdb, file = "data/GO/catdb.RData")
批量进行GO富集分析
这段代码主要用于进行批量的基因本体(Gene Ontology, GO)富集分析,首先根据DEG结果定义DEG集合,然后执行批量的GO富集分析和GO slim富集分析。最后,将结果保存在相应的变量中。具体步骤如下:
-
载入所需的包和数据:通过加载"biomaRt"包和预处理好的基因到GO的CATdb对象(从之前的代码段中加载)。同时,也加载了之前进行差异表达分析得到的DEG结果(DEG_list)。
-
定义DEG集合:根据DEG结果,将上调和下调基因分别命名为"名称_up_down"、"名称_up"和"名称_down"的命名空间。创建DEG集合(DEGlist)将这些命名空间组合起来,并移除长度为0的集合。
-
执行批量的GO富集分析:使用
GOCluster_Report函数对DEG集合进行批量的GO富集分析。设置方法参数(method)为"all",表示返回所有通过设定的p-value阈值(cutoff)的GO术语。指定id_type参数为"gene",表示基因标识符类型。还可以设置其他参数,如聚类阈值(CLSZ)、GO分类(gocats)和记录指定的GO术语(recordSpecGO)。将结果保存在BatchResult变量中。 -
获取GO slim向量:通过使用"biomaRt"包获取特定生物体的GO slim向量。选择合适的BioMart数据库(“plants_mart”)和数据集(“athaliana_eg_gene”),然后使用
getBM函数获取"goslim_goa_accession"属性并将结果转换为字符向量。 -
执行GO slim富集分析:使用
GOCluster_Report函数对DEG集合进行GO slim富集分析。设置方法参数(method)为"slim",表示仅返回在"goslimvec"向量中指定的GO术语。其他参数的设置与批量GO富集分析类似。将结果保存在BatchResultslim变量中。
## 6.2 Batch GO term enrichment analysis
## Apply the enrichment analysis to the DEG sets obtained the above differential
## expression analysis. Note, in the following example the FDR filter is set here to an
## unreasonably high value, simply because of the small size of the toy data set used ## in
## this vignette. Batch enrichment analysis of many gene sets is performed with the
## function. When method=all, it returns all GO terms passing the p-value cutoff
## specified under the cutoff arguments. When method=slim, it returns only the GO
## terms specified under the myslimv argument. The given example shows how a GO
## slim vector for a specific organism can be obtained from BioMart.
library("biomaRt")
load("data/GO/catdb.RData")
DEG_list <- filterDEGs(degDF = edgeDF, filter = c(Fold = 2, FDR = 50),
plot = FALSE)
up_down <- DEG_list$UporDown
names(up_down) <- paste(names(up_down), "_up_down", sep = "")
up <- DEG_list$Up
names(up) <- paste(names(up), "_up", sep = "")
down <- DEG_list$Down
names(down) <- paste(names(down), "_down", sep = "")
DEGlist <- c(up_down, up, down)
DEGlist <- DEGlist[sapply(DEGlist, length) > 0]
BatchResult <- GOCluster_Report(catdb = catdb, setlist = DEGlist,
method = "all", id_type = "gene", CLSZ = 2, cutoff = 0.9,
gocats = c("MF", "BP", "CC"), recordSpecGO = NULL)
library("biomaRt")
m <- useMart("plants_mart", dataset = "athaliana_eg_gene", host = "https://plants.ensembl.org")
goslimvec <- as.character(getBM(attributes = c("goslim_goa_accession"),
mart = m)[, 1])
BatchResultslim <- GOCluster_Report(catdb = catdb, setlist = DEGlist,
method = "slim", id_type = "gene", myslimv = goslimvec, CLSZ = 10,
cutoff = 0.01, gocats = c("MF", "BP", "CC"), recordSpecGO = NULL)
绘制批量GO富集分析的结果
使用goBarplot函数绘制批量GO富集分析结果的条形图。首先选择感兴趣的子集,然后分别绘制不同GO类别的条形图。具体步骤如下:
-
子集选择:首先从BatchResultslim数据框中选择与"M6-V6_up_down"匹配的行,将结果存储在gos变量中。然后将整个BatchResultslim数据框存储在gos变量中,以便后续绘制。
-
绘制MF(分子功能)类别的GO条形图:通过调用
goBarplot函数绘制MF类别的GO条形图。将gos作为输入数据框,并设置gocat参数为"MF"。将结果保存为PDF文件。 -
绘制BP(生物过程)类别的GO条形图:通过再次调用
goBarplot函数绘制BP类别的GO条形图。将gos作为输入数据框,并设置gocat参数为"BP"。 -
绘制CC(细胞组分)类别的GO条形图:通过再次调用
goBarplot函数绘制CC类别的GO条形图。将gos作为输入数据框,并设置gocat参数为"CC"。
## 6.3 Plot batch GO term results
## The data.frame generated by GOCluster can be plotted with the goBarplot
## function. Because of the variable size of the sample sets, it may not always be
## desirable to show the results from different DEG sets in the same bar plot. Plotting
## single sample sets is achieved by subsetting the input data frame as shown in the
## first line of the following example.
gos <- BatchResultslim[grep("M6-V6_up_down", BatchResultslim$CLID),
]
gos <- BatchResultslim
pdf("GOslimbarplotMF.pdf", height = 8, width = 10)
goBarplot(gos, gocat = "MF")
dev.off()
goBarplot(gos, gocat = "BP")
goBarplot(gos, gocat = "CC")
这里以分子功能为例。
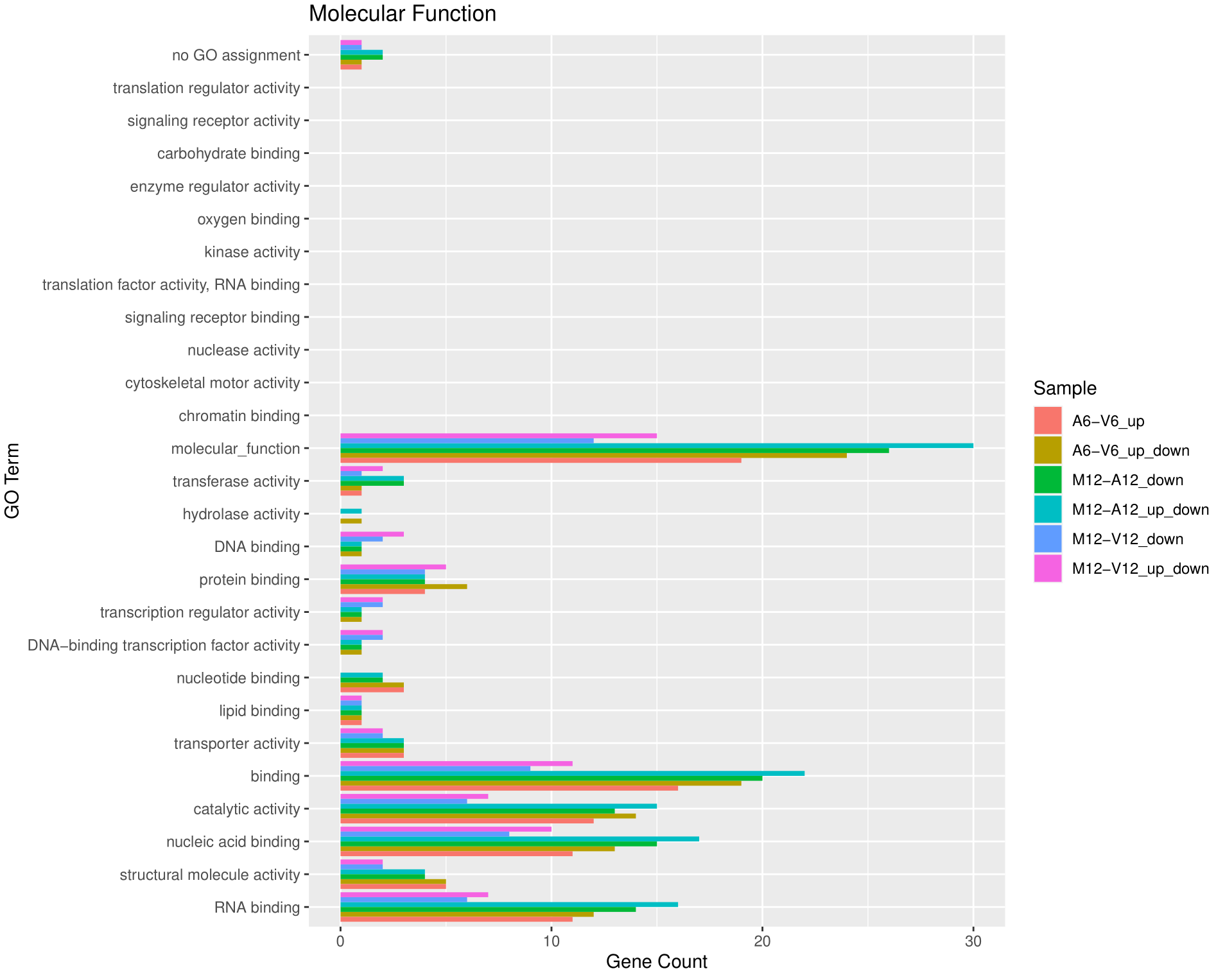
层次聚类和热图绘制
使用pheatmap库进行层次聚类和热图绘制。首先提取感兴趣基因的表达矩阵子集,然后基于Pearson相关系数计算距离并进行层次聚类,最后绘制热图以可视化聚类结果。具体步骤如下:
-
导入必要的库:通过加载pheatmap库,准备进行层次聚类和热图分析。
-
提取差异表达基因(DEGs)的表达矩阵:从上述差异表达分析中确定的DEGs中提取基因的rlog转换后的表达矩阵。这里使用了DEG_list[[1]]作为DEGs的标识,通过将其转换为字符向量并去除重复项,得到基因的唯一标识符。
-
提取感兴趣基因的表达值:从rlog转换后的表达矩阵中,提取与感兴趣基因的唯一标识符相对应的行,得到一个子集矩阵y。
-
绘制热图:通过调用pheatmap函数绘制热图。设置scale参数为"row",对行进行标准化;设置clustering_distance_rows参数和clustering_distance_cols参数为"correlation",使用基于Pearson相关系数的距离度量进行行和列的层次聚类。将结果保存为PDF文件。
## 7. Clustering and heat maps
#####################################################
## The following example performs hierarchical clustering on the rlog transformed
## expression matrix subsetted by the DEGs identified in the above differential
## expression analysis. It uses a Pearson correlation-based distance measure and
## complete linkage for cluster joining.
library(pheatmap)
geneids <- unique(as.character(unlist(DEG_list[[1]])))
y <- assay(rlog(dds))[geneids, ]
pdf("heatmap1.pdf")
pheatmap(y, scale = "row", clustering_distance_rows = "correlation",
clustering_distance_cols = "correlation")
dev.off()
![RDEKA[3V5VGK4]DURRNWX%F](https://img-blog.csdnimg.cn/img_convert/7a092792ec5001deef5ee8315da29363.png)