大家好,我是邓飞,现在写博客越来越繁琐了,每个平台对图片都有自己的规则,不能通用,各种找不到图片,本着充值是我变强的原则,买了Markdown Nice的VIP(https://product.mdnice.com/),据说实现了一键发布多个平台,而且有自己的图库,今天先水一篇,试试效果。
星球上有老师问:snpEFF和bedtools都可以注释基因,他们有什么区别?
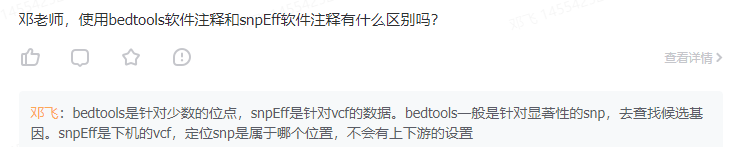
当我无法立即回答时,我都会说:“这是一个好问题”,然后就开始找答案或者总结答案,当时我的回答:
bedtools是针对少数的位点,snpEff是针对vcf的数据。bedtools一般是针对显著性的snp,去查找候选基因。snpEff是下机的vcf,定位snp是属于哪个位置,不会有上下游的设置
上面的回答基本解释了两者的差异,但是,因为要水一篇,所以要解释一下写篇博客!
1. bedtools和snpEFF软件介绍
这两款软件,官网上详细介绍都有:
bedtools:
https://bedtools.readthedocs.io/en/latest/index.html
snpEFF:
http://pcingola.github.io/SnpEff/
简短的来说,bedtools可以处理基因型数据,各种复杂的功能,匹配、删减、格式转换等,称为生信界的瑞士军刀。
snpEFF好像专注于基因注释。
2. 区别1:数据格式不一样
vcftools需要gff文件和bed数据,才可以进行注释。
snpEFF,输入文件是vcf格式,另外,他需要基因组数据和gff创建数据库(通用的物种官网有现成的,但是推荐自定义构建,不容易出错。
3. 区别2:能否指定上下游区间
snpEFF不能指定上下游区间,精确的给出位点在基因组上的区域(内含子、外显子、哪个基因等等)
bedtools,可以指定上下游区间,对于GWAS分析得到的显著性位点,因为存在LD,所以更灵活,应用更多。
4. 区别3:应用领域
snpEFF,主要是下机数据,vcf数据,运算速度快,给出每个SNP的信息
bedtools,主要是需要设置上下游区间的位点,比如GWAS得到的显著性位点。
上面就是两者的区别,bedtools注释基因的写过好几篇博客了,下一篇介绍snpEFF注释vcf的教程。

![[深度学习]yolov7 pytorch模型转onnx,转ncnn模型和mnn模型使用细节](https://img-blog.csdnimg.cn/fd2628d6ecbf45eca14ed7c0d096c7e3.png#pic_center)