 clc;
clc;
clear all;
close all
warning off
tic
%% 导入数据
% 训练集——190个样本
P_train = xlsread('data','training set','B2:G191')';
T_train= xlsread('data','training set','H2:H191')';
% 测试集——44个样本
P_test=xlsread('data','test set','B2:G45')';
T_test=xlsread('data','test set','H2:H45')';
N = size(P_test, 2); % 测试集样本数
M = size(P_train, 2); % 训练集样本数
%% 数据归一化
%% 数据归一化
[p_train, ps_input] = mapminmax(P_train, 0, 1);
p_test = mapminmax('apply', P_test, ps_input);
[t_train, ps_output] = mapminmax(T_train, 0, 1);
t_test = mapminmax('apply', T_test, ps_output);
%构建网络
%% 节点个数
inputnum=6;
hiddennum=8;
outputnum=1;
net=newff(p_train,t_train,hiddennum);%单隐含层,5个隐含层神经元
%% 遗传算法参数初始化
maxgen=20; %进化代数,即迭代次数
sizepop=10; %种群规模
pcross=0.2; %交叉概率选择,0和1之间
pmutation=0.1; %变异概率选择,0和1之间
%节点总数:输入隐含层权值、隐含阈值、隐含输出层权值、输出阈值(4个基因组成一条染色体)
numsum=inputnum*hiddennum+hiddennum+hiddennum*outputnum+outputnum;%21个,10,5,5,1
lenchrom=ones(1,numsum);%个体长度,暂时先理解为染色体长度,是1行numsum列的矩阵
bound=[-3*ones(numsum,1) 3*ones(numsum,1)]; %是numsum行2列的串联矩阵,第1列是-3,第2列是3
%------------------------------------------------------种群初始化--------------------------------------------------------
individuals=struct('fitness',zeros(1,sizepop), 'chrom',[]); %将种群信息定义为一个结构体:10个个体的适应度值,10条染色体编码信息
avgfitness=[]; %每一代种群的平均适应度,一维
bestfitness=[]; %每一代种群的最佳适应度
bestchrom=[]; %适应度最好的染色体,储存基因信息
%初始化种群
for i=1:sizepop
%随机产生一个种群
individuals.chrom(i,:)=Code(lenchrom,bound); %编码(binary(二进制)和grey的编码结果为一个实数,float的编码结果为一个实数向量)
x=individuals.chrom(i,:);
%计算适应度
individuals.fitness(i)=fun(x,inputnum,hiddennum,outputnum,net,p_train,t_train); %染色体的适应度
end
FitRecord=[];
%找最好的染色体
[bestfitness, bestindex]=min(individuals.fitness);%bestindex是最小值的索引(位置/某个个体),bestfitness的值为最小适应度值
bestchrom=individuals.chrom(bestindex,:); %最好的染色体,从10个个体中挑选到的
avgfitness=sum(individuals.fitness)/sizepop; %染色体的平均适应度(所有个体适应度和 / 个体数)
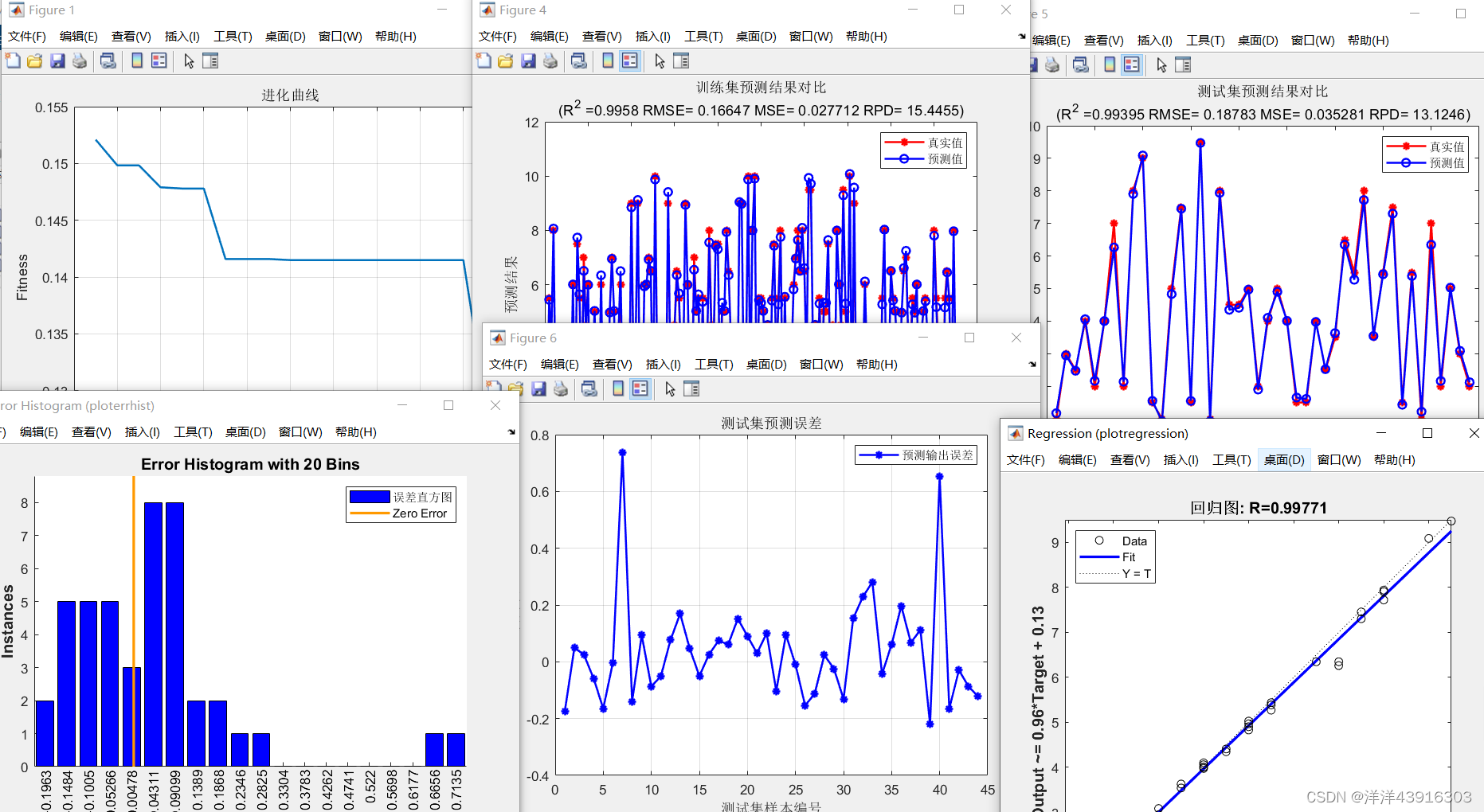
% 记录每一代进化中最好的适应度和平均适应度
trace=[avgfitness bestfitness]; %trace矩阵,1行2列,avgfitness和bestfitness仅仅是数值
![[附源码]计算机毕业设计springboot美发店会员管理系统](https://img-blog.csdnimg.cn/81725abc55e54714918b3ea58b835a35.png)


![[附源码]计算机毕业设计JAVA校园快递管理系统](https://img-blog.csdnimg.cn/1c129fa6d5eb411fa0be419bde6c0b90.png)




![[附源码]计算机毕业设计JAVA闲置物品线上交易系统](https://img-blog.csdnimg.cn/bbd4bc35d2be4b5a9d7097e291a67326.png)

![L. Paid Leave(贪心)[CCPC Finals 2021]](https://img-blog.csdnimg.cn/6506b439fac14d668f9eadd877d23f9d.png)