文章目录
- 写在前面
- FusionMap融合检测原理
- FusionMap与其他软比较
- FusionMap分析流程
- FusionMap结果文件说明
- FusionMap mono CUP设置
图片来源: https://en.wikipedia.org/wiki/Fusion_gene
写在前面
下面主要内容是关于RNA-seq数据分析融合,用到软件是FusionMap 【FusionMap参考文献】。
融合分析使用哪个软件,哪个软件表现较好,在Biostarts发现一个问答列举了一些软件(看这里),里面有STAR-Fusion, STAR-Fusion, deFuse, FusionCatcher等30多个融合分析软件,其中约20多个软件的文献发表于2011-2013年,FusionMap软件的文献也发表与2011年。还有几篇软件比较的文献,各分析软件的优劣文献中也会提,晚一些发表的文献也会与之前发表的软件作比较。
另外,FusionMap软件应该很早不再更新了,是在Oshell工具包中进行维护。
FusionMap融合检测原理
融合Reads:Seed reads和Rescued reads
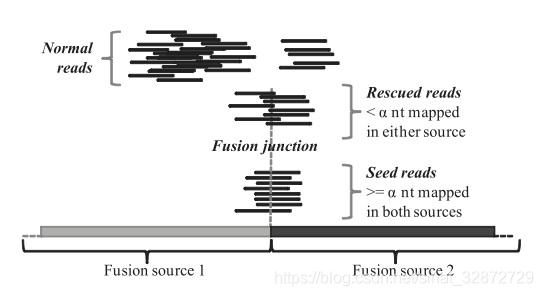
融合方向:图来源
FusionMap与其他软比较

图片来源文献: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4797269/
上面表格红色标注是,相对FusionMap, 结果比FusionMap差的。从该文献给的上表中粗略看,FusionMap在三组和构造的数据集上表现还可以,而在乳腺癌和黑色素瘤的样本数据上表现较差,对它的综合评价属于中等程度,但它有个最大的好处,就是用C#编写,其运行速度较其他软件要快。
对于该软比较文献中,具体使用的是什么样的数据,各软件分析时使用的参数,比较的评分标准,可能对各个软件都会有影响。
FusionMap分析流程
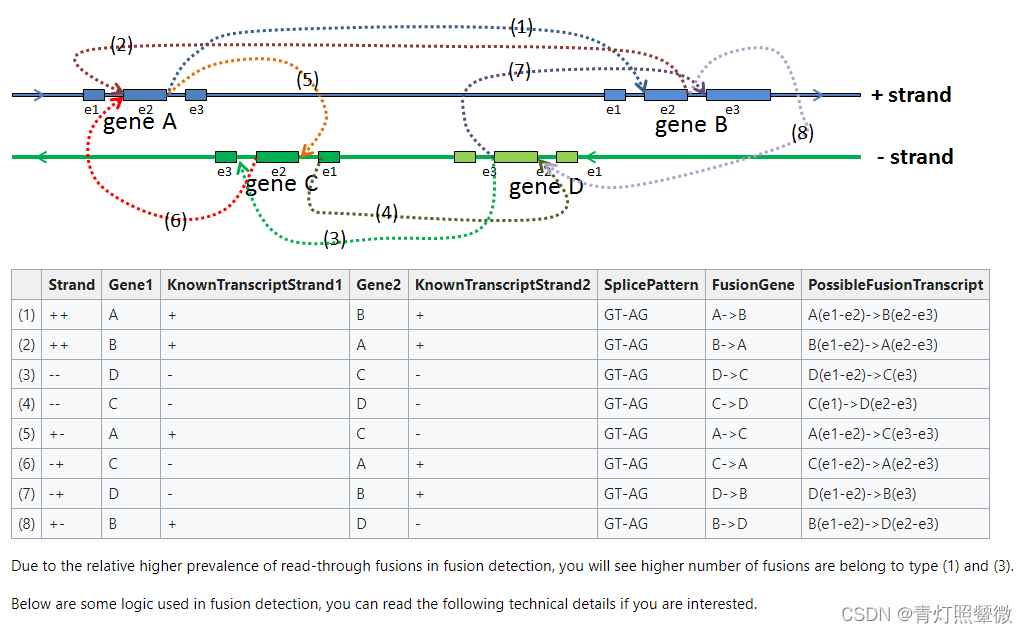
软件分析流程pipline:图来源

融合检测流程:

其中,序列比对是在GSNAP软件基础上进行了一些改进。介绍GSNAP
(1)分析流程配置oscript文件示例:
http://www.arrayserver.com/wiki/index.php?title=OmicScript_example_for_RNA-Seq_data_analysis_pipeline
(2)软件使用示例:
mono oshell.exe --runscript Base_Dir Script_path/buildIndex.oscript Temp_Dir Mono_Path
FusionMap结果文件说明
结果文件report:http://www.arrayserver.com/wiki/index.php?title=Fusion_SE_report
| 表头名称 | 含义 |
|---|---|
| FusionID | 融合ID信息,格式为: FUS_Start_END 注 [ 1 ] ^{注[1]} 注[1] |
| Bam.UniqueCuttingPositionCount | Uniq read数,相当于Seed Reads+Rescued reads的去重 |
| Bam.SeedCount | 如上图中,假设
α
α
α就是一端softclip长度最小值,则SeedCount则为softclip长度>=α的Reads数。(如果值比较小,也可能根本比不上)目的是这些Reads可作为种子序列扩展成较长的融合序列,再将扩展的融合序列作为自构建ref,比较靠边缘的融合序列比对到自构建ref。如果是PE150bp,
α
=
25
α=25
α=25,最多可扩展成125+125=250bp的融合序列 |
| Bam.RescuedCount | 相当于softclip长度<α的reads数,通过SeedReads自构建的ref进行比对上的reads |
| Strand | 链方向 |
| Chromosome1 | 断点1染色体 |
| Position1 | 断点1位置 |
| Chromosome2 | 断点2染色体 |
| Position2 | 断点2位置 |
| KnownGene1 | 断点1基因 |
| KnownTranscript1 | 断点1转录本 |
| KnownExonNumber1 | 断点1外显子号 |
| KnownTranscriptStrand1 | 断点1基因链方向 |
| KnownGene2 | 断点2基因 |
| KnownTranscript2 | 断点2转录本 |
| KnownExonNumber2 | 断点2外显子号 |
| KnownTranscriptStrand2 | 断点2基因链方向 |
| FusionJunctionSequence | 融合断点上下游(30bp)序列 |
| FusionGene | 融合两端基因 |
| SplicePattern | 融合剪接模式 [1] |
| SplicePatternClass | 融合剪接模式类型 [1] |
| FrameShift | 发生frameshift的格式 [2] |
| FrameShiftClass | frameshift的类型 [2] |
| Distance | 融合断点间距离(不是同一染色体时为-1) |
| OnExonBoundary | 是否在Exon边界,None:两个断点都不在;Both:两个断点都在;Single:有一个断点在。 |
| Filter | 可过滤信息,包括:InFamilyList(家族基因列表)/InBlackList(黑名单列表) |
其中:
[1] SplicePatternClass包括:
- CanonicalPatter[Major]: GT-AG
SplicePattern - CanonicalPatter[Minor]: GC-AG and AT-AC
SplicePattern - NonCanonicalPatter: all other detected di-nucleotides
[2] FrameShiftClass包括:
- FrameShift:融合处发生了移码。
- InFrame:融合处是整码(断点处基因的碱基是3的倍数)。
FrameShift对应值的格式为: [0{0,1}1{0,1}2{0,1}->0{0,1}1{0,1}2{0,1}(python正则表达式,0{0,1}是指0字符出现0-1次)
- (1)若值为
->或->0{0,1}1{0,1}2{0,1}或0{0,1}1{0,1}2{0,1}->(例如,->0/->01/->012),可能是融合比对的位置两个断点或一端断点不在编码区?【InFrame】 - (2)若值为
0->1或1->2或2->0(下图),表示两个基因融合后,没有发生移码【InFrame】 - (3)若值为
0->2/0->0、1->0/1->1或2->1/2->2,表示两个基因融合后,发生了移码 【FrameShift】 - (4)若值为
->左端或右端有多种模式,例如02->2,012->1。当多种模式都包含在情形(3)中,则为【FrameShift】,例如:0->02、01->0、02->2、1->01、12->1、2->12;当多种模式中至少有一种属于情形(2),则为【InFrame】,例如:0->012、0->01、01->01等。
图来源 也介绍了fusionMap检测融合:

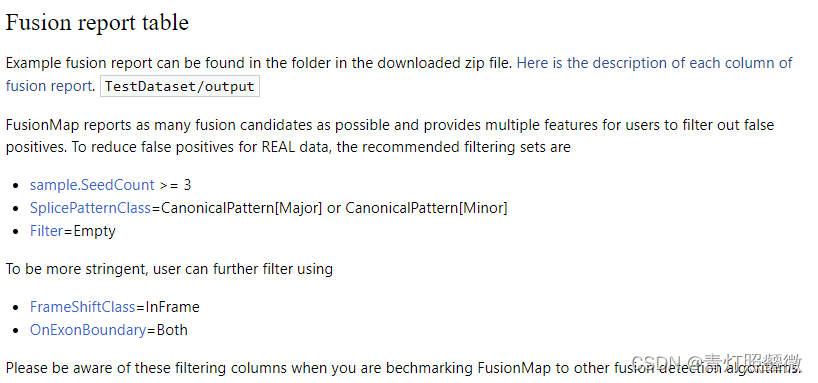
推荐的过滤条件:图来源
SeedCount >= 3; SplicePatternClass =CanonicalPattern[Major] or CanonicalPattern[Minor] ; Filter=Empty
更严格的条件:FrameShiftClass=InFrame;OnExonBoundary=Both
单端/双端融合的基因表达结果:图来源(oscript配置中设定分析表达步骤)

FusionMap mono CUP设置
占用较高CPU问题,如何设置?【还不清楚】
-
FusionMap使用说明文档
-
关于FusionMap的一些安装说明中提到控制文档示例中,有提到mono的一些参数设置:(但不知道在哪里设置该文件?)

Mono的帮助文档中有相关参数:
| 参数 | 说明 |
|---|---|
| –aot | |
| 环境变量: | |
| MONO_CPU_ARCH | 覆盖自动 CPU 检测机制。目前仅用于arm, eg:MONO_CPU_ARCH="armv4 thumb" mono ... |
| MONO_THREADS_PER_CPU | 一般线程池中的最大线程数将为 20 + (MONO_THREADS_PER_CPU * CPU 数)。此变量的默认值为 10 |
| MONO_TLS_SESSION_CACHE_TIMEOUT | SSL/TLS 会话缓存将保留其条目以避免客户端和服务器之间的新协商的时间(以秒为单位)。协商非常占用 CPU,因此特定于应用程序的自定义值可能证明对小型嵌入式系统有用。默认值为 180 秒。 |
其他参考:
- github上mono相关问题
MONO_THREADS_PER_CPU=100参考 - 在linux上使用mono跑c#的程序一定要特别注意,while(true)的问题(使用sleep)参考
- 配置supervisor来管理mono程序 参考
- https://www.mono-project.com/docs/
- 融合发生机制及检测方法