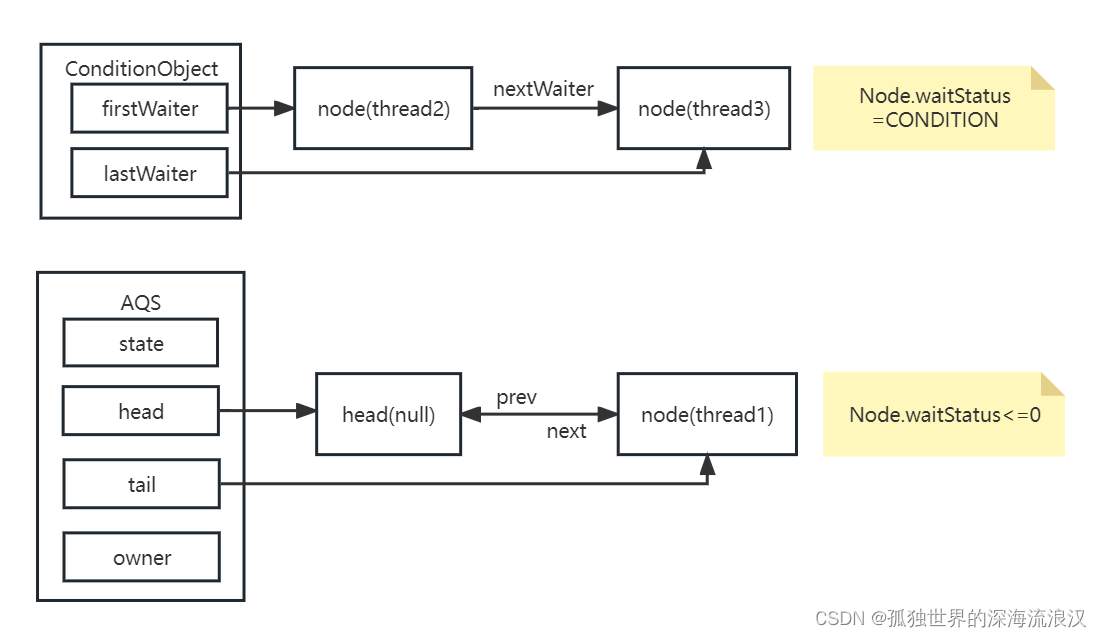
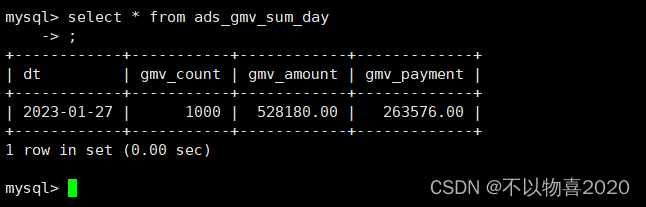
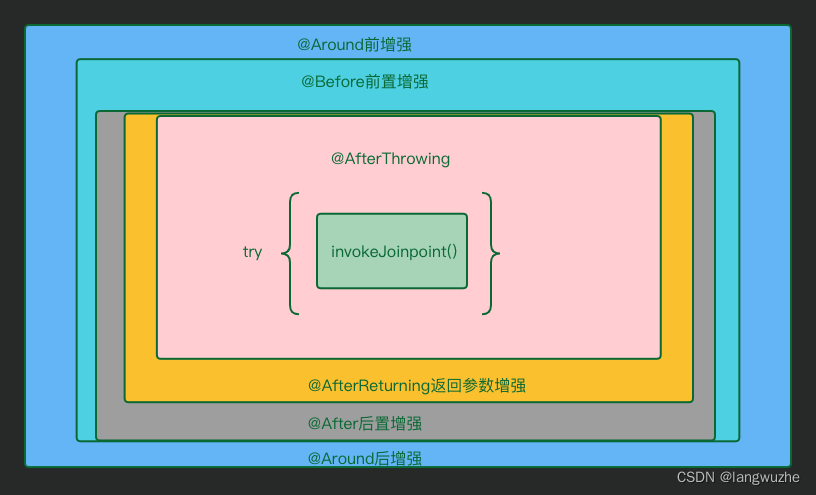
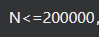1. 切割位点
ATACseq 应该在较小的保护区(如转录因子结合位点)周围生成较短的片段(我们的无核小体区域)。
因此,我们可以在不同组织/细胞类型/样本中寻找围绕感兴趣基序的切割位点堆积。
为了从我们的 BAM 文件中生成切割位点,我们首先将读取大小调整为 1bp,并根据链进行 4/-5 bp 的偏移,以调整插入 Tn5 转座酶的预期偏移。
在这里,我们将识别通过任意截止的 CTCF 基序,然后使用 soGGi 在它们周围绘制切割位点。我们将跳回我们的 Greenleaf 数据集来执行此操作。
2. 查找 motifs
我们需要确定 CTCF 基序在基因组中的位置,因此首先我们需要知道 CTCF 基序是什么样的。
motifDB 包包含来自公共数据库(例如 JASPAR)的有关 Motif 的信息。在这里,我们使用带有我们感兴趣的主题 (CTCF) 的 query() 函数来提取 CTCF 主题。
library(MotifDb)
library(Biostrings)
library(BSgenome.Hsapiens.UCSC.hg19)
CTCF <- query(MotifDb, c("CTCF"))
CTCF
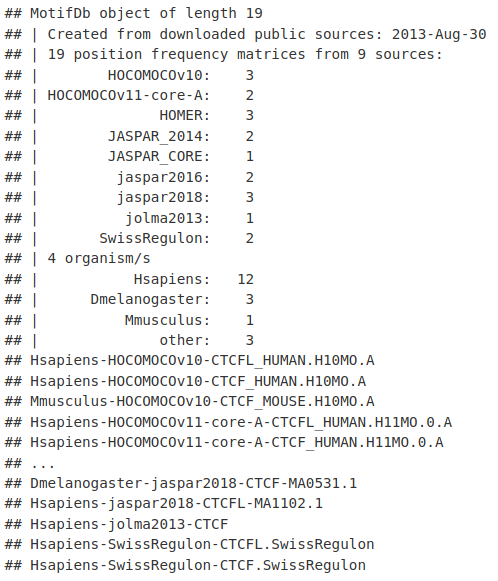
我们可以为 CTCF 提取一个点权重矩阵,它指定了 DNA 碱基出现在 CTCF 基序中的可能性。在这里,我们从 Human JASPAR Core 数据库中提取 CTCF 的主题。
names(CTCF)
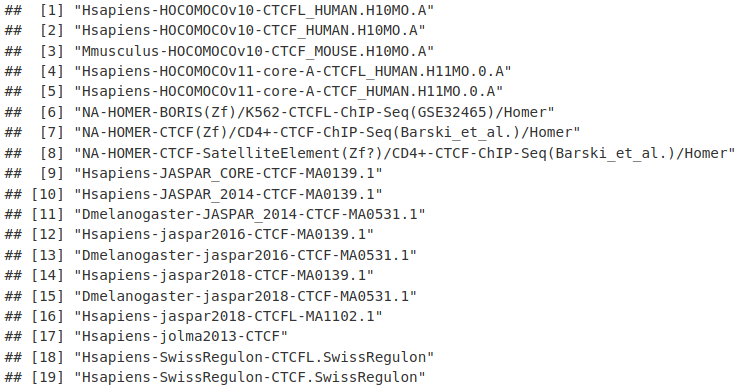
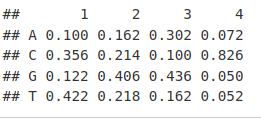
ctcfMotif <- CTCF[[1]]
ctcfMotif[, 1:4]
3. PWMs 可视化
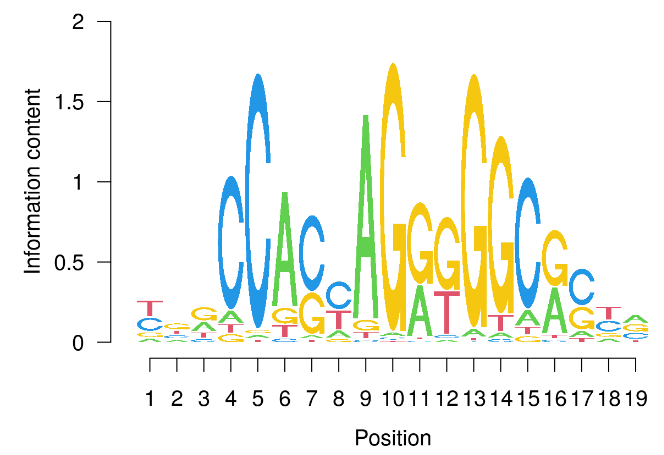
我们可以使用 seqLogo 包和 seqLogo 函数可视化主题中 DNA 碱基的频率。
library(seqLogo)
seqLogo(ctcfMotif)
4. PWMs 搜索
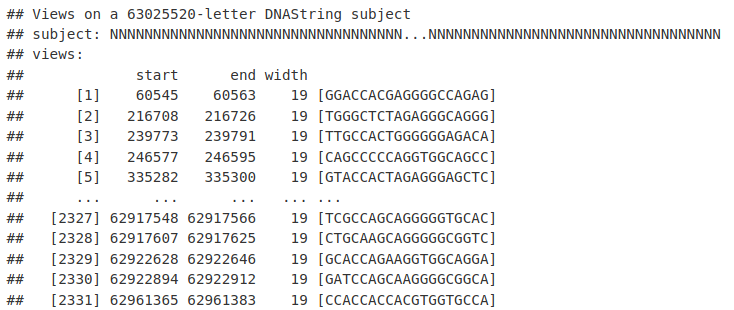
我们现在可以将 matchPWM() 函数与我们新获得的 CTCF PWM 一起使用。在这里,我们将使用 BSgenome 库中为人类 BSgenome.Hsapiens.UCSC.hg19 提供的序列搜索 Chr20 上的序列。结果是一个 Views 对象,类似于 IRanges 对象。
myRes <- matchPWM(ctcfMotif, BSgenome.Hsapiens.UCSC.hg19[["chr20"]])
myRes
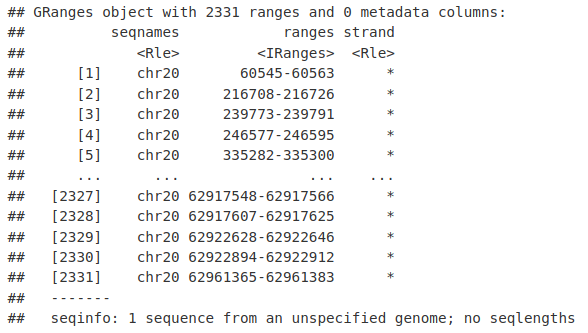
我们需要将 Views 对象转换为 GRanges,以便我们可以在 soGGi 中使用它们来绘制切割站点。
toCompare <- GRanges("chr20", ranges(myRes))
toCompare
5. 切割位点分析
要绘制切割位点,我们希望只考虑读取的 5' 端,并且需要调整已知的 5' 读取偏移量到实际 T5 切割位点。
这将涉及捕获读数的 5' 端并将正链和负链上的读数分别移动 4bp 或 -5bp。
首先,我们读入我们的无核小体区域 BAM 文件并提取读取对。
BAM <- "~/Downloads/ATAC_Workshop/ATAC_Data/ATAC_BAM/Sorted_ATAC_50K_2_openRegions.bam"
atacReads_Open <- readGAlignmentPairs(BAM)
read1 <- first(atacReads_Open)
read2 <- second(atacReads_Open)
read2[1, ]

现在我们可以根据链将两个读取对的 5' 端移动 4bp 或 -5bp。这从两个读数中产生了我们所有切割位点的 GRanges。
Firsts <- resize(granges(read1), fix = "start", 1)
First_Pos_toCut <- shift(granges(Firsts[strand(read1) == "+"]), 4)
First_Neg_toCut <- shift(granges(Firsts[strand(read1) == "-"]), -5)
Seconds <- resize(granges(read2), fix = "start", 1)
Second_Pos_toCut <- shift(granges(Seconds[strand(read2) == "+"]), 4)
Second_Neg_toCut <- shift(granges(Seconds[strand(read2) == "-"]), -5)
test_toCut <- c(First_Pos_toCut, First_Neg_toCut, Second_Pos_toCut, Second_Neg_toCut)
test_toCut[1:2, ]

现在我们可以使用 coverage() 函数使用切割位点位置的 GRanges 生成整个基因组切割位点的 RLElist。
cutsCoverage <- coverage(test_toCut)
cutsCoverage20 <- cutsCoverage["chr20"]
cutsCoverage20[[1]]

我们可以使用带有 soGGi 的 RLElist 围绕我们发现的 CTCF 图案生成切割位点图。
我们将格式更改为 rlelist,并将 distanceAround 参数更改为 500bp。
CTCF_Cuts_open <- regionPlot(cutsCoverage20, testRanges = toCompare, style = "point",
format = "rlelist", distanceAround = 500)
现在我们可以使用 plotRegion() 函数绘制切割点。
plotRegion(CTCF_Cuts_open, outliers = 0.001) + ggtitle("NucFree Cuts Centred on CTCF") +
theme_bw()

本文由 mdnice 多平台发布