1写在前面
今天是医师节,祝各位医护节日快乐,夜班平安,病历全是甲级,没有错误。🥰
不知道各位医师节的福利是什么!?😂
我们医院是搞了义诊活动,哈哈哈哈哈哈哈。🫠
这么好的节日,以后尽量别搞了。😭
2用到的包
rm(list = ls())
library(slingshot)
library(tidyverse)
library(uwot)
library(mclust)
library(RColorBrewer)
library(grDevices)
3示例数据
means <- rbind(
matrix(rep(rep(c(0.1,0.5,1,2,3), each = 300),100),
ncol = 300, byrow = T),
matrix(rep(exp(atan( ((300:1)-200)/50 )),50), ncol = 300, byrow = T),
matrix(rep(exp(atan( ((300:1)-100)/50 )),50), ncol = 300, byrow = T),
matrix(rep(exp(atan( ((1:300)-100)/50 )),50), ncol = 300, byrow = T),
matrix(rep(exp(atan( ((1:300)-200)/50 )),50), ncol = 300, byrow = T),
matrix(rep(exp(atan( c((1:100)/33, rep(3,100), (100:1)/33) )),50),
ncol = 300, byrow = TRUE)
)
counts <- apply(means,2,function(cell_means){
total <- rnbinom(1, mu = 7500, size = 4)
rmultinom(1, total, cell_means)
})
rownames(counts) <- paste0('G',1:750)
colnames(counts) <- paste0('c',1:300)
sce <- SingleCellExperiment(assays = List(counts = counts))
sce

4基因过滤
这里我们把cluster size设置为≥10,count设置为≥3,以这个条件进行过滤,筛选过一些低表达的。😏
geneFilter <- apply(assays(sce)$counts,1,function(x){
sum(x >= 3) >= 10
})
sce <- sce[geneFilter, ]
5Normalization
接着我们做一下Normalization,去除一些技术因素或者生物学因素上的影响,比如batch,sequencing depth, cell cycle等等。🥰
FQnorm <- function(counts){
rk <- apply(counts,2,rank,ties.method='min')
counts.sort <- apply(counts,2,sort)
refdist <- apply(counts.sort,1,median)
norm <- apply(rk,2,function(r){ refdist[r] })
rownames(norm) <- rownames(counts)
return(norm)
}
assays(sce)$norm <- FQnorm(assays(sce)$counts)
6降维
这里我们用2种方法来试试,PCA和UMAP。😂
6.1 方法一
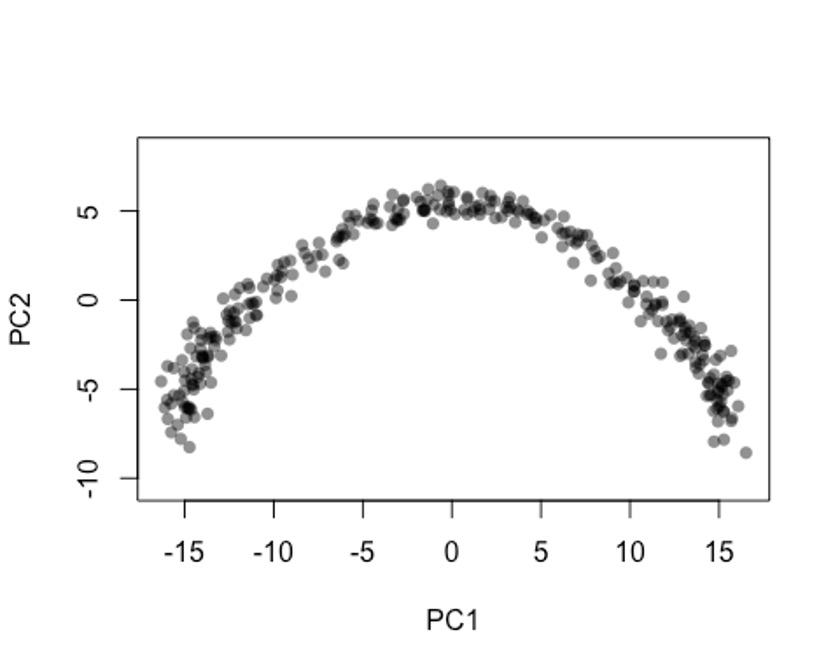
在做PCA的时候,大家尽量不要对基因的表达量做scale,这样的结果会更准一些。🤐
pca <- prcomp(t(log1p(assays(sce)$norm)), scale. = F)
rd1 <- pca$x[,1:2]
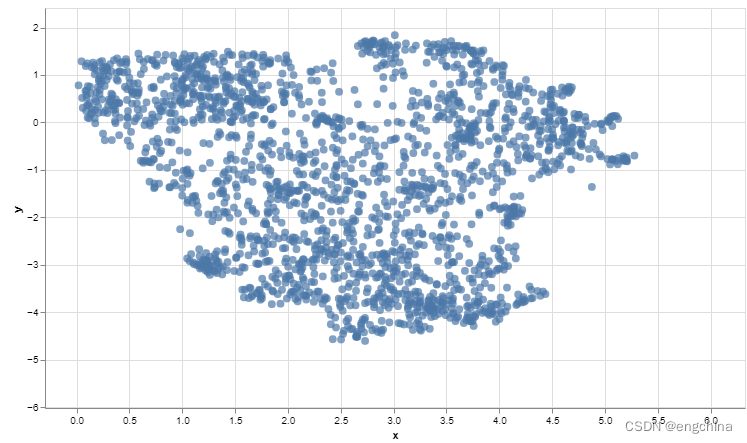
plot(rd1, col = rgb(0,0,0,.5), pch=16, asp = 1)
6.2 方法二
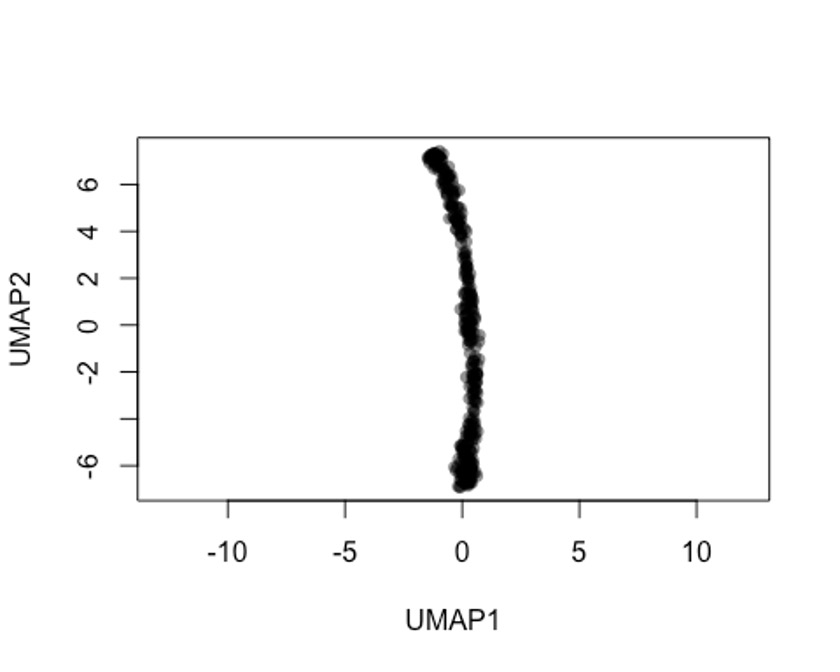
rd2 <- uwot::umap(t(log1p(assays(sce)$norm)))
colnames(rd2) <- c('UMAP1', 'UMAP2')
plot(rd2, col = rgb(0,0,0,.5), pch=16, asp = 1)
然后我们把结果存在到之前的SingleCellExperiment文件sce里吧。😜
reducedDims(sce) <- SimpleList(PCA = rd1, UMAP = rd2)
7细胞聚类
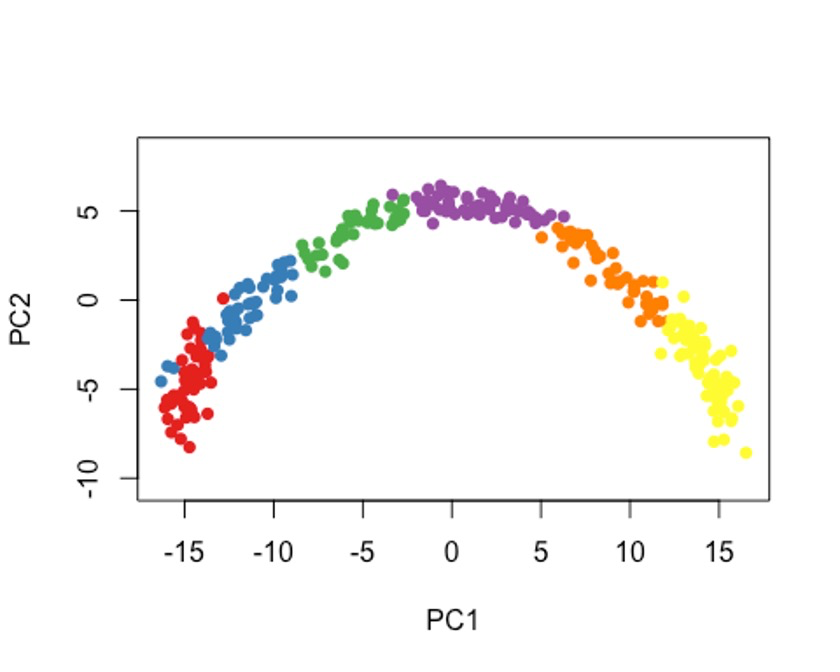
这里我们也提供2种方法吧,Gaussian mixture modeling和k-means。🥳
7.1 方法一
cl1 <- Mclust(rd1)$classification
colData(sce)$GMM <- cl1
plot(rd1, col = brewer.pal(9,"Set1")[cl1], pch=16, asp = 1)
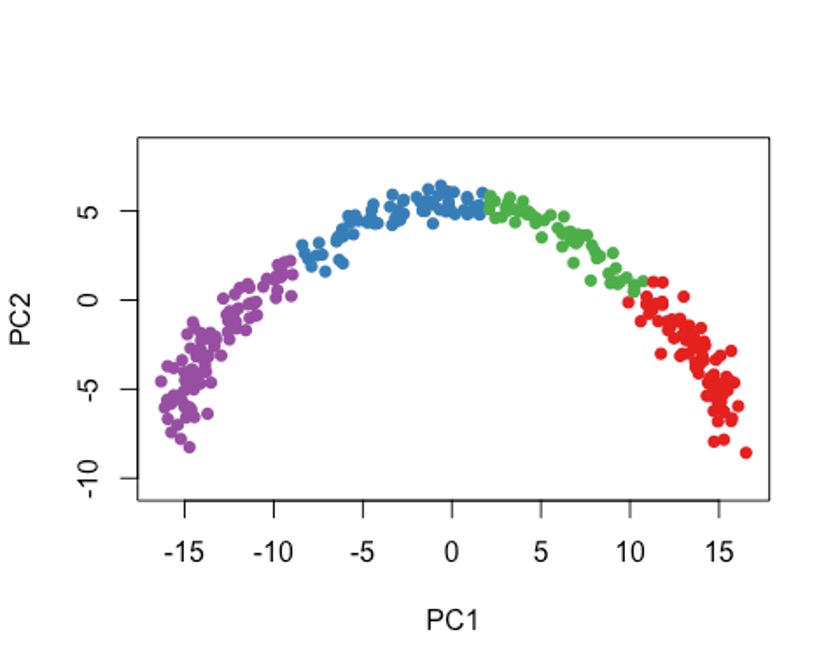
7.2 方法二
cl2 <- kmeans(rd1, centers = 4)$cluster
colData(sce)$kmeans <- cl2
plot(rd1, col = brewer.pal(9,"Set1")[cl2], pch=16, asp = 1)
8slingshot进行Pseudotime分析
sce <- slingshot(sce, clusterLabels = 'GMM', reducedDim = 'PCA')
summary(sce$slingPseudotime_1)

colData(sce)$slingshot

single-trajectory 🤩
colors <- colorRampPalette(brewer.pal(11,'Spectral')[-6])(100)
plotcol <- colors[cut(sce$slingPseudotime_1, breaks=100)]
plot(reducedDims(sce)$PCA, col = plotcol, pch=16, asp = 1)
lines(SlingshotDataSet(sce), lwd=2, col='black')
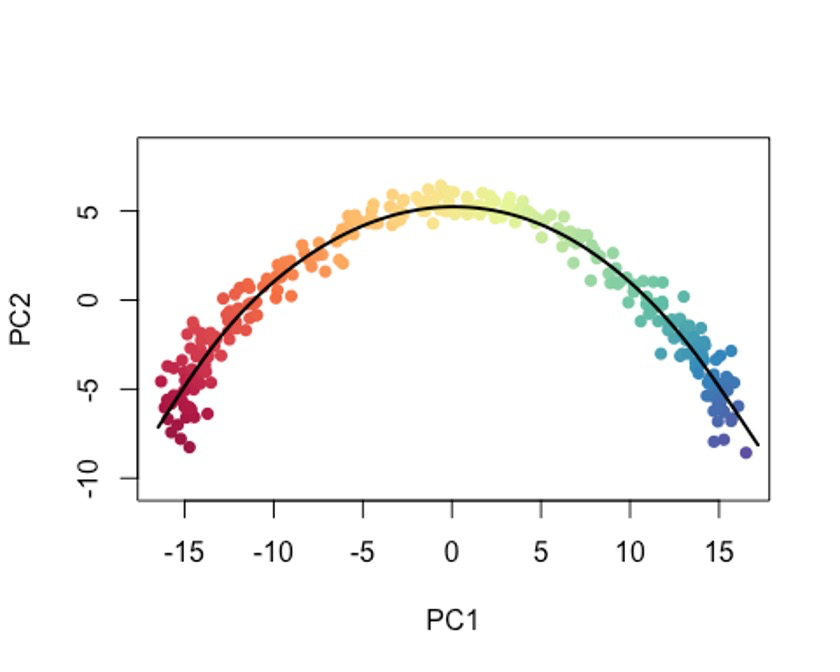
当然,你也可以把type设置为lineages,模拟以cluster为单位的变化过程。🤓
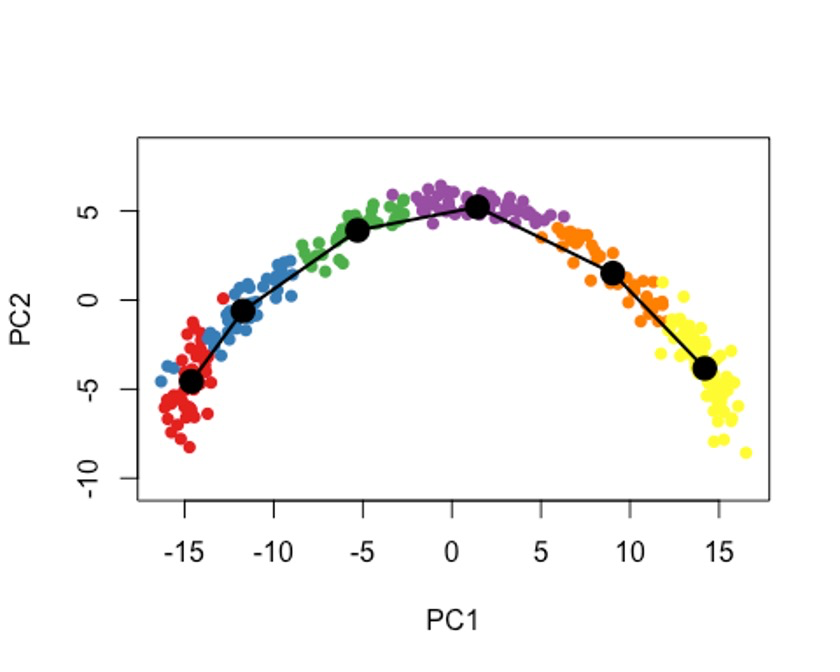
plot(reducedDims(sce)$PCA, col = brewer.pal(9,'Set1')[sce$GMM], pch=16, asp = 1)
lines(SlingshotDataSet(sce), lwd=2, type = 'lineages', col = 'black')
9大数据的处理
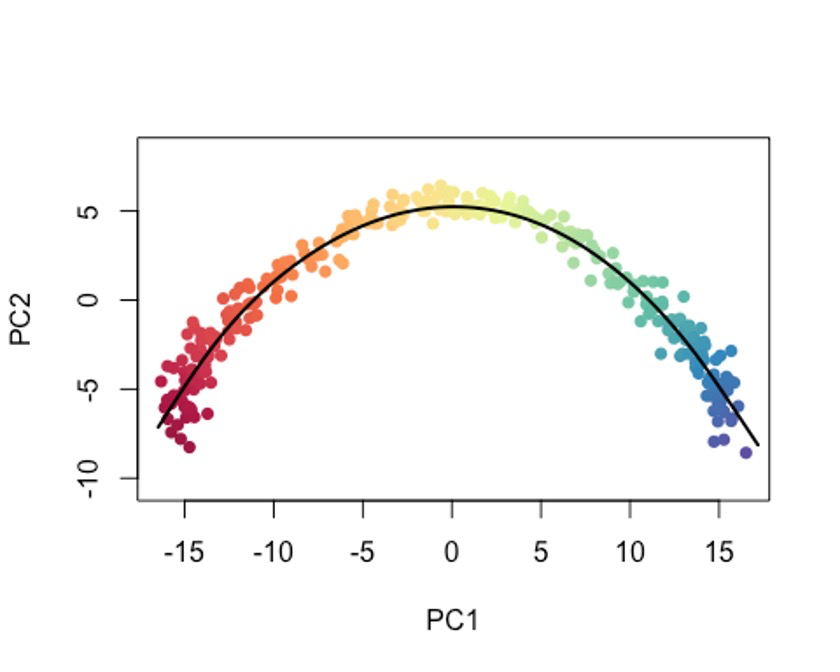
示例数据都比较小,但实际你测完的数据可能会非常大。😜
这个时候我们要加上一个参数,approx_points,默认是150,一般选择100-200。🫠
如果你把approx_points设置为False,会获得尽可能多的点,和data中的细胞数有一定的关系。😭
sce5 <- slingshot(sce, clusterLabels = 'GMM', reducedDim = 'PCA',
approx_points = 150)
colors <- colorRampPalette(brewer.pal(11,'Spectral')[-6])(100)
plotcol <- colors[cut(sce5$slingPseudotime_1, breaks=100)]
plot(reducedDims(sce5)$PCA, col = plotcol, pch=16, asp = 1)
lines(SlingshotDataSet(sce5), lwd=2, col='black')
10将细胞投射到现有轨迹上
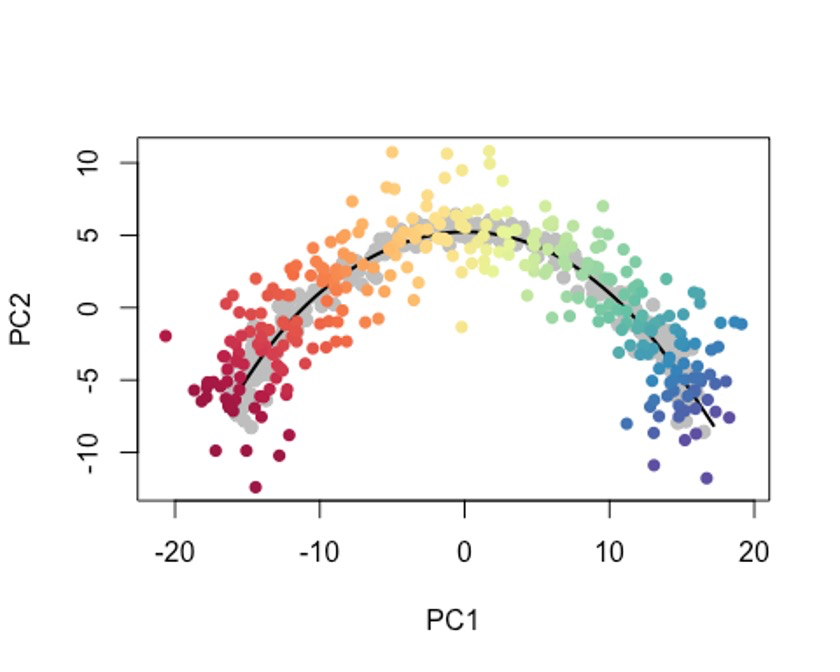
可能有时候,我们只想使用细胞的一个子集或者新的data来确定轨迹,我们都需要一种方法来确定新细胞沿着先前构建的轨迹的位置。🤓
这个时候我们可以用predict函数,
pto <- sce$slingshot
newPCA <- reducedDim(sce, 'PCA') + rnorm(2*ncol(sce), sd = 2)
newPTO <- slingshot::predict(pto, newPCA)
原细胞轨迹为灰色哦。🫠
newplotcol <- colors[cut(slingPseudotime(newPTO)[,1], breaks=100)]
plot(reducedDims(sce)$PCA, col = 'grey', bg = 'grey', pch=21, asp = 1,
xlim = range(newPCA[,1]), ylim = range(newPCA[,2]))
lines(SlingshotDataSet(sce), lwd=2, col = 'black')
points(slingReducedDim(newPTO), col = newplotcol, pch = 16)

点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 🤩 LASSO | 不来看看怎么美化你的LASSO结果吗!?
📍 🤣 chatPDF | 别再自己读文献了!让chatGPT来帮你读吧!~
📍 🤩 WGCNA | 值得你深入学习的生信分析方法!~
📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码!
📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程
📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~
📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~
📍 🧐 rms | 批量完成你的线性回归
📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图
📍 🤠 Network | 高颜值动态网络可视化工具
📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图
📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程)
📍 ......



本文由 mdnice 多平台发布