VET:Vcf Export Tools
工具简介
VET是一个基于R语言开发的变异位点信息批量提取工具,主要功能是根据VCF数据集,按照基因ID、样品ID、变异位点ID等参数,实现批量提取,同时支持变异位点结构注释,一步搞定变异数据的快速提取。
########## WelCome to VCF Export Tools ###########
>>>>>>>>>>>>>>>> Design By BioNote <<<<<<<<<<<<<<<<<
可选参数:
[1]根据基因ID提取变异数据
[2]根据物理位置提取变异数据
[3]根据样品名称提取变异数据
[4]根据SNP名称提取变异数据
--------------------------------------------------
[INFO]第一个参数填选项,第二个参数填项目备注名称
[INFO]第三个参数选择是否过滤样本,默认为“N”
[INFO]第四个参数选择是否进行结构注释,默认为“N”
[INFO]运行方法 $ Rscript ./run.R 1 test Y Y
>>>>>>>>>>>>>>>> 程序版本:V 2.0.1 <<<<<<<<<<<<<<<<
##################################################
功能与应用
基因测序后经过上游分析得到的VCF文件储存了所有样本对应的所有变异信息,通常数据量非常大,在实际使用中需要根据情况对指定信息进行提取。目前已有vcftools或bcftools等工具能够实现上述操作,但是用的时候参数比较复杂,整个过程略显繁琐。
本工具集成了R、tidyverse、Python、vcftools、bcftools、snpEff等常用工具,开发便捷式流程实现批量操作。
主要应用场景是对大规模VCF原始数据集进行提取,支持多种个性化方式:
-
按基因ID提取指定基因内变异信息 -
按材料名称提取某些材料变异信息 -
按变异位点名称筛选指定变异信息 -
按照物理位置提取指定区段内数据
支持流式操作,提取后筛选指定样品并对每个变异位点进行结构注释(判断错义突变、移码突变等),最终将结果文件打包生成压缩包。
使用方法
-
第一步:输入待提取的信息 -
第二步:运行Run.R脚本 -
第三步:下载结果文件
可以批量操作,无需手写代码。
原理介绍
VCF是生信研究中储存基因变异信息的重要格式,通常经过测序上游分析后得到一份具有丰富信息量的vcf或者vcf.gz文件。

以“##”开头的行表示注释信息,一般记录了字段类型和历史命令,这部分相当于一个日志信息。剩下的数据部分类似一个表格,大体上每行是一个变异位点,每列是一个材料样本。
变异位点(简称SNP)是按照染色体上不同位置进行统计的,展示不同材料中在某个位置碱基差异。
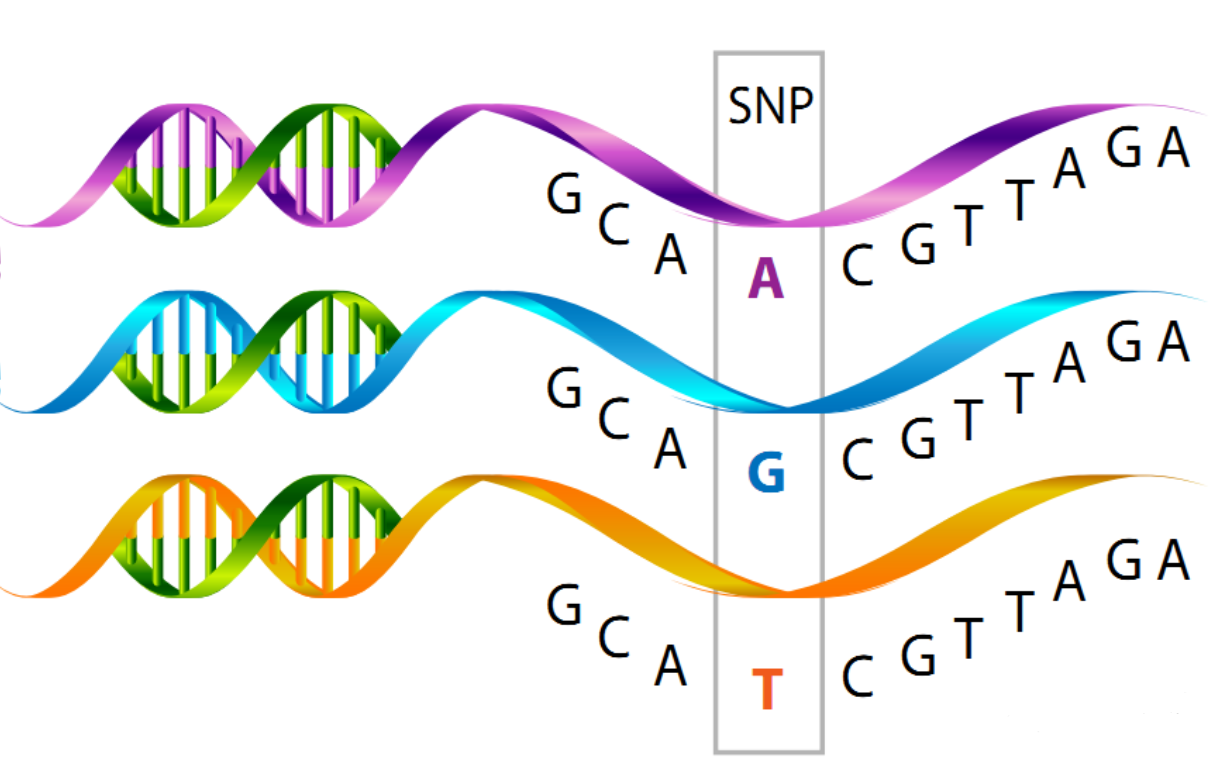
按照突变的类型可以分为3种类型:
-
缺失:Del,某些碱基不见了 -
插入:Ins,新出现了某些碱基 -
替换:SNP,单核苷酸多态性变异
 一般常见的VCF文件主要由上述信息组成,对于大规模测序得到的多个样品合并VCF文件,可能包含几千万行×几千列(亿级数据量)。
一般常见的VCF文件主要由上述信息组成,对于大规模测序得到的多个样品合并VCF文件,可能包含几千万行×几千列(亿级数据量)。
在实际进行分析时,可能只需要考虑某几个基因或者是一小段区间内的变异数据,因此需要对VCF文件进行提取,只取出想要的一小部分,这个过程涉及到Linux下不同软件的相互配合。
VET 源代码
首先,建立项目文件夹并生成以下结构:
Aug 9 16:30 00_scripts
Aug 17 15:24 01_INPUT_GeneID.txt
Aug 17 16:09 01_out_byGeneID
Aug 9 18:30 02_INPUT_Postion.txt
Aug 10 11:25 02_out_byPostion
Aug 10 11:02 03_INPUT_SampleName.txt
Aug 4 15:13 03_out_bySampleName
Aug 4 15:31 04_INPUT_SNP.txt
Aug 4 15:14 04_out_bySNP
Aug 6 11:34 05_INPUT_filevcf.txt
Aug 6 11:33 05_out_bySnpEff
程序初始化
下面的代码为程序初始化过程,将会加载tidyverse等软件包,并读取重要参数,完成后将获得输出提示。
#!/usr/local/bin/Rscript
# VCF Export Tools 基因型变异数据批量提取工具,快捷提取VCF文件
# 依赖软件:Python、bcftools、tidyverse、snpeff
suppressPackageStartupMessages(library("cli"))
suppressPackageStartupMessages(library("tidyverse"))
cli::cli_text("########## WelCome to VCF Export Tools ###########
\n >>>>>>>>>>>>>>>> Design By Jewel <<<<<<<<<<<<<<<<<
\n可选参数:
\n\t[1]根据基因ID提取变异数据
\n\t[2]根据物理位置提取变异数据
\n\t[3]根据样品名称提取变异数据
\n\t[4]根据SNP名称提取变异数据
\n--------------------------------------------------
\n[INFO]第一个参数填选项,第二个参数填项目备注名称
\n[INFO]第三个参数选择是否过滤样本,Y为过滤指定样本
\n[INFO]第四个参数为'Y'时将对vcf文件进行变异结构注释
\n[INFO]例如 $ ./run.R 1 test Y Y
\n>>>>>>>>>>>>>>>> 程序版本:V 2.0.1 <<<<<<<<<<<<<<<<
\n ##################################################")
args <- commandArgs(T)
if(length(args)!=4){stop("参数输入有误,请检查输入格式,示例“./Run.R 1 jobname Y/N Y/N")}
# CONFIG SETTING:
db_file <- "wgs_all.vcf.gz" # 设置数据库名称
db_name <- "WGS"
# 程序初始化,删除上次输出结果文件----------
OPT <- args[1] # 程序子选项
JOB <- args[2] # 项目备注信息
SAM <- args[3] # 是否过滤样本
EFF <- args[4] # 是否结构注释
print(str_c("INFO 当前选择的数据库为:",db_file))
print(str_c("INFO 当前项目名称为: ",OPT," <-> ",JOB))
print(str_c("INFO 是否对样品进行过滤(Y为过滤指定样本,否则不过滤):"),SAM)
print(str_c("INFO 是否对vcf进行结构变异注释(Y为进行注释,否则不注释):"),EFF)
system("rm -rf ./01_out_byGeneID/*")
system("rm -rf ./02_out_byPostion/*")
system("rm -rf ./03_out_bySampleName/*")
system("rm -rf ./04_out_bySNP/*")
system("rm -rf ./05_out_bySnpEff/*")
cli::cli_text("INFO 系统输出文件夹初始化完成")
根据基因ID提取变异信息
根据输入的参数进行判断,如果选项为1,则执行下面的步骤,主要调用Python程序进行信息检索,并由bcftools工具批量提取变异信息,若需要根据指定样品进行过滤,则利用view功能对样品进行筛选,最后生成结果压缩文件。
if (OPT == "1"){
cli::cli_text("INFO 待提取的基因ID如下,将自动自取上下游3000bp内的变异数据")
id <- read.table("./01_INPUT_GeneID.txt",header = F)
print(id$V1)
cli::cli_text("INFO 基因ID信息整理完毕,接下来开始检索物理区间")
system("Rscript prefix_gene_filter.R ./01_INPUT_GeneID.txt")
cli::cli_text("INFO 接下来执行Python脚本调用bcftools提取基因变异信息")
system(str_c("python bcftools_view_filiter_Chr.py --input ./00_scripts/id.txt --vcf ./",
db_file))
cli::cli_text("INFO 提取完成,对结果进行打包压缩")
if (SAM == "Y"){
for (i in 1:nrow(id)){
system(str_c("bcftools view --force-samples -S ",
"./03_INPUT_SampleName.txt ",
id$V1[i],".vcf.gz > ",
id$V1[i],".vcf"))
}
system("mv ./Traes*vcf ./01_out_byGeneID/")
system("rm -rf ./Traes*vcf.gz")
system(str_c("tar -czvf ",format(Sys.Date(), "%Y_%m_%d"),"_",JOB,"_ExportFrom_",db_name,
"_LOTSample_Filter_ByGeneID",".tar.gz ./01_out_byGeneID/* ./Tips.pdf"))
}else{
system("mv ./Traes* ./01_out_byGeneID/")
system(str_c("tar -czvf ",format(Sys.Date(), "%Y_%m_%d"),"_",JOB,"_ExportFrom_",db_name,
"_AllSample_Filter_ByGeneID",".tar.gz ./01_out_byGeneID/* ./Tips.pdf"))
}
cli::cli_text("INFO 任务运行结束,请及时下载结果文件,下次运行前将清空结果文件")
}
根据物理位置提取变异信息
根据指定的物理区间判断染色体的和起止位置,并结果VCF文件筛选指定区间内的变异数据,采用bcftools的 filter功能进行实现,提取完成后进行打包压缩。
if (OPT == "2"){
cli::cli_text("INFO 待提取物理区间如下,正在提取中......")
region <- read.table("./02_INPUT_Postion.txt",header = F)
for (i in 1:nrow(region)){
print(str_c("Index: ",i," Region: ",region$V1[i]," Info: ",region$V2[i]))
system(str_c("bcftools filter ",db_file," --regions ",region$V1[i]," > ",region$V1[i],"_",region$V2[i],".vcf"))
system("mv Chr* ./02_out_byPostion/")
system("rename : _ ./02_out_byPostion/*")
system("rename - _ ./02_out_byPostion/*")
}
cli::cli_text("INFO 提取完成,对结果进行打包压缩")
system(str_c("tar -czvf ",format(Sys.Date(), "%Y_%m_%d"),"_",JOB,"_ExportFrom_",db_name,
"_AllSample_Filter_ByPositin",".tar.gz ./02_out_byPostion/* ./Tips.pdf"))
cli::cli_text("INFO 任务运行结束,请及时下载结果文件,下次运行前将清空结果文件")
}
今天分享的内容就到这里,还有两个功能正在开发中,之后有时间再分享关于提取指定位点名称和结构注释的方法,目前这项工具还未完成,如需抢先体验请后台联系,后续将开源至Github,欢迎转发分享。
参考资料:
https://mp.weixin.qq.com/s/DdXyqiW7c7lCp4103flQZQ
本文由 mdnice 多平台发布