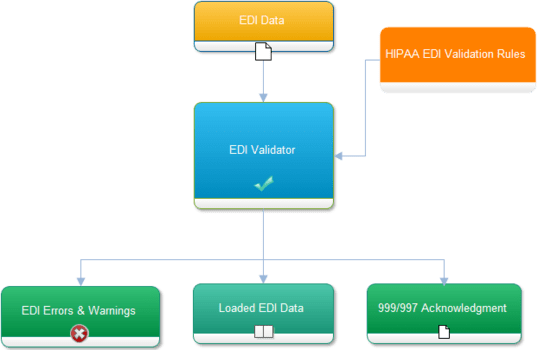
AlphaFold2源码解析(8)–模型之三维坐标构建

这个模块我们讲解AlphaFold的Structure module模块,该结构模块将蛋白质结构的抽象表示映射为具体的三维原子坐标。 Evoformer的单一表征被用作初始单一表征
s
i
i
n
i
t
i
a
l
{s^{initial}_i }
siinitial,
s
i
i
n
i
t
i
a
l
∈
R
s
c
s^{initial}_i \in R^c_s
siinitial∈Rsc,而Evoformer的对表征
z
i
j
{z_{ij}}
zij,
z
i
j
∈
R
c
z
z_{ij} \in R^{c_z}
zij∈Rcz 并且
i
,
j
∈
{
1
,
.
.
.
,
N
r
e
s
}
i,j \in \{1,...,N_{res}\}
i,j∈{1,...,Nres}在注意操作中偏重亲和图。该模块有8个具有共享权重的层。 每一层都会更新抽象的单一表征
s
i
{s_i}
si以及具体的三维表征(“残留物气体”),它被编码为每个残留物
T
i
{T_i}
Ti的一个骨架。 我们通过一个元组
T
i
:
=
(
R
i
,
t
⃗
i
)
T_i:= (R_i,\vec{t}_i)
Ti:=(Ri,ti)来表示frame。该元组表示从本地frame到全局参考frame的欧几里得变换。也就是说,它将本地坐标中的位置
x
⃗
l
o
c
a
l
∈
R
3
\vec{x}_{local} \in R^3
xlocal∈R3转换为全球坐标中的位置
x
⃗
g
l
o
b
a
l
∈
R
3
\vec{x}_{global} \in R^3
xglobal∈R3,即

在具有旋转矩阵和平移矢量的参数化中,这就是:
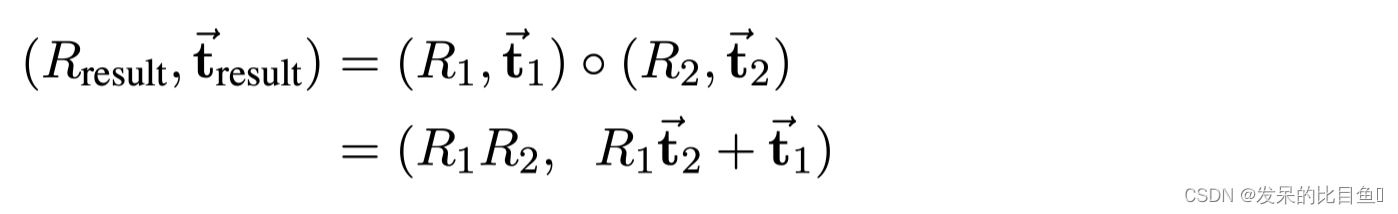
为了获得所有的原子坐标,我们通过扭转角对每个残基进行参数化。也就是说,扭角是唯一的自由度,而所有的键角和键长都是完全刚性的。 各个原子用它们的名字来识别
S
a
t
o
m
n
a
m
e
s
=
{
N
,
C
α
,
C
,
O
,
C
β
,
C
γ
,
C
γ
1
,
C
γ
2
,
.
.
.
.
}
S_{atom names}= \{N, C^α, C, O, C^β, C^γ, C^{γ1}, C^{γ2}, . ... \}
Satomnames={N,Cα,C,O,Cβ,Cγ,Cγ1,Cγ2,....}。 扭转被命名为
S
t
o
r
s
i
o
n
n
a
m
e
s
=
{
ω
,
φ
,
ψ
,
χ
1
,
χ
2
,
χ
3
,
χ
4
}
S_{torsion names} = \{ω, φ, ψ, χ^1, χ^2, χ^3, χ^4\}
Storsionnames={ω,φ,ψ,χ1,χ2,χ3,χ4}。我们根据原子对扭力角的依赖性将其分为 “刚性组”。
一个浅层ResNet预测了扭转角 α i f ⃗ ∈ R 2 \vec{\alpha^f_i}\in R^2 αif∈R2。每当它们被用作角度时,它们会通过归一化被映射到单位圆上的点。 此外,我们引入了一个小的辅助损失,鼓励原始向量的单位准则,以避免退化值。与[0, 2π]角度表示法相比,这种表示法没有不连续性,可以直接用于构建旋转矩阵,而不需要三角函数。预测的扭转角被转换为刚性原子组的frame。
在训练过程中,每层的最后一步是计算当前三维结构的辅助损失。中间的FAPE损失只对骨干框架和 C α C^α Cα原子位置进行操作,以保持低计算成本。出于同样的原因,侧链在这里只由它们的扭角来监督。一些刚性组的180◦旋转对称性也通过提供替代角度 α ⃗ i a l t t r u t h , f \vec{\alpha}_i^{alt truth,f} αialttruth,f来解决。
我们发现将刚体的方向分量的梯度归零是很有帮助的,所以任何迭代都是为了在当前迭代中找到结构的最佳方向,但并不关心是否有一个更适合下一次迭代的方向。 从经验上看,这提高了训练的稳定性,大概是通过消除连锁组成框架中产生的杠杆效应。在8层之后,最终的骨干框架和扭角被映射到所有刚性基团(骨干和侧链) T i f T^f_i Tif和所有原子坐标 x ⃗ i a \vec{x}^a_i xia的框架上。
在训练过程中,预测的框架和原子坐标通过FAPE损失与地面真相进行比较,该损失评估了所有原子坐标(骨架和侧链)相对于所有刚性基团的情况。 少数刚性基团的180◦旋转对称性是通过对地面真实结构中的模糊原子进行全局一致的重命名来处理的。
最后,该模型以每个残基的预测 l D D T − C α lDDT-Cα lDDT−Cα得分(pLDDT)的形式预测其置信度。这个分数是根据预测结构和真实结构计算出来的每个残基的真实lDDT-Cα分数来训练的。
下图是AlphaFold论文中“Highly accurate protein structure prediction with AlphaFold”关于结构模块流程,
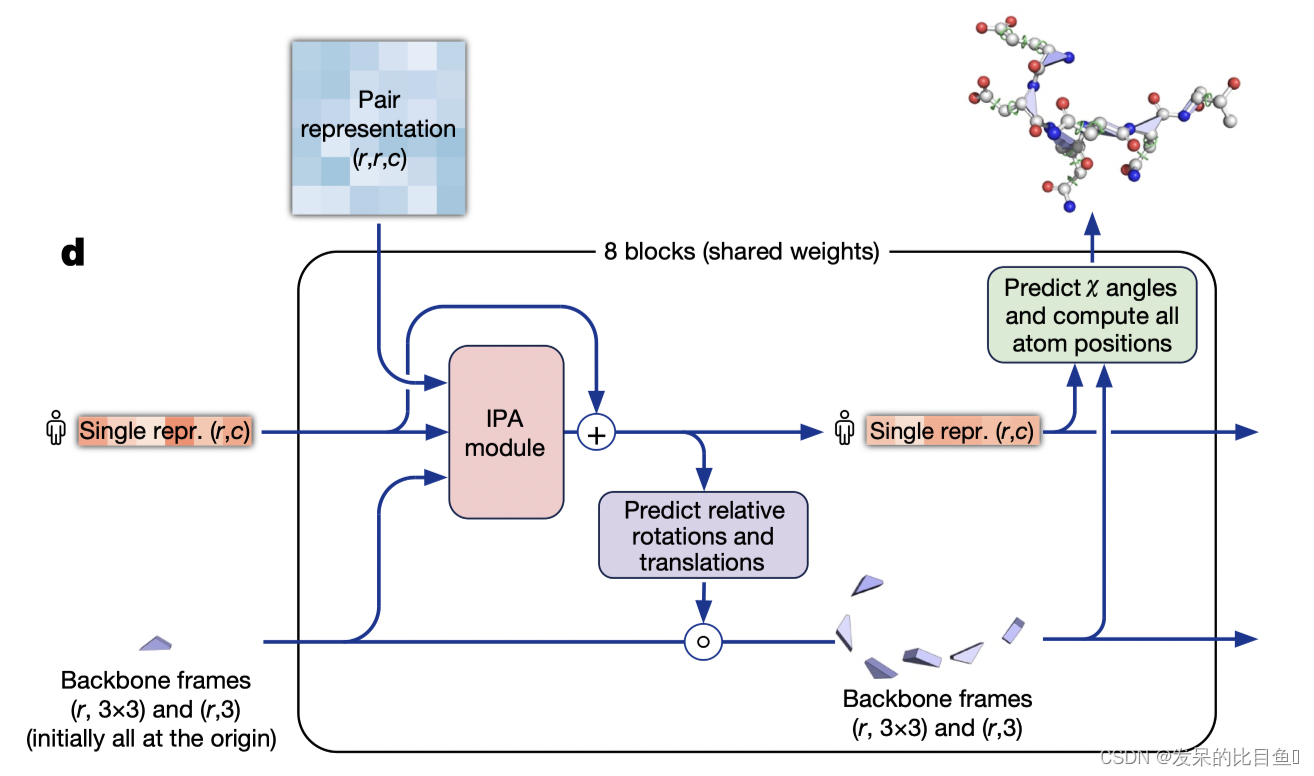
这个模块在补充材料有具体为代码说明:

包含了几个模块:
从真实原子结构构建frame
我们使用来自真实PDB结构的三个原子的位置,通过Gram-Schmidt过程构建框架, 注意,平移矢量
t
⃗
\vec{t}
t被分配给中心原子
x
2
⃗
\vec{x_2}
x2。对于骨干框架,我们用N作为
x
⃗
1
\vec{x}_1
x1,Cα作为
x
⃗
2
\vec{x}_2
x2,C作为
x
⃗
3
\vec{x}_3
x3,所以框架的中心是Cα。对于侧链框架,我们使用扭键之前的原子作为
x
⃗
1
\vec{x}_1
x1,扭键之后的原子作为
x
⃗
2
\vec{x}_2
x2,之后的下一个原子作为
x
⃗
3
\vec{x}_3
x3。

IPA模块
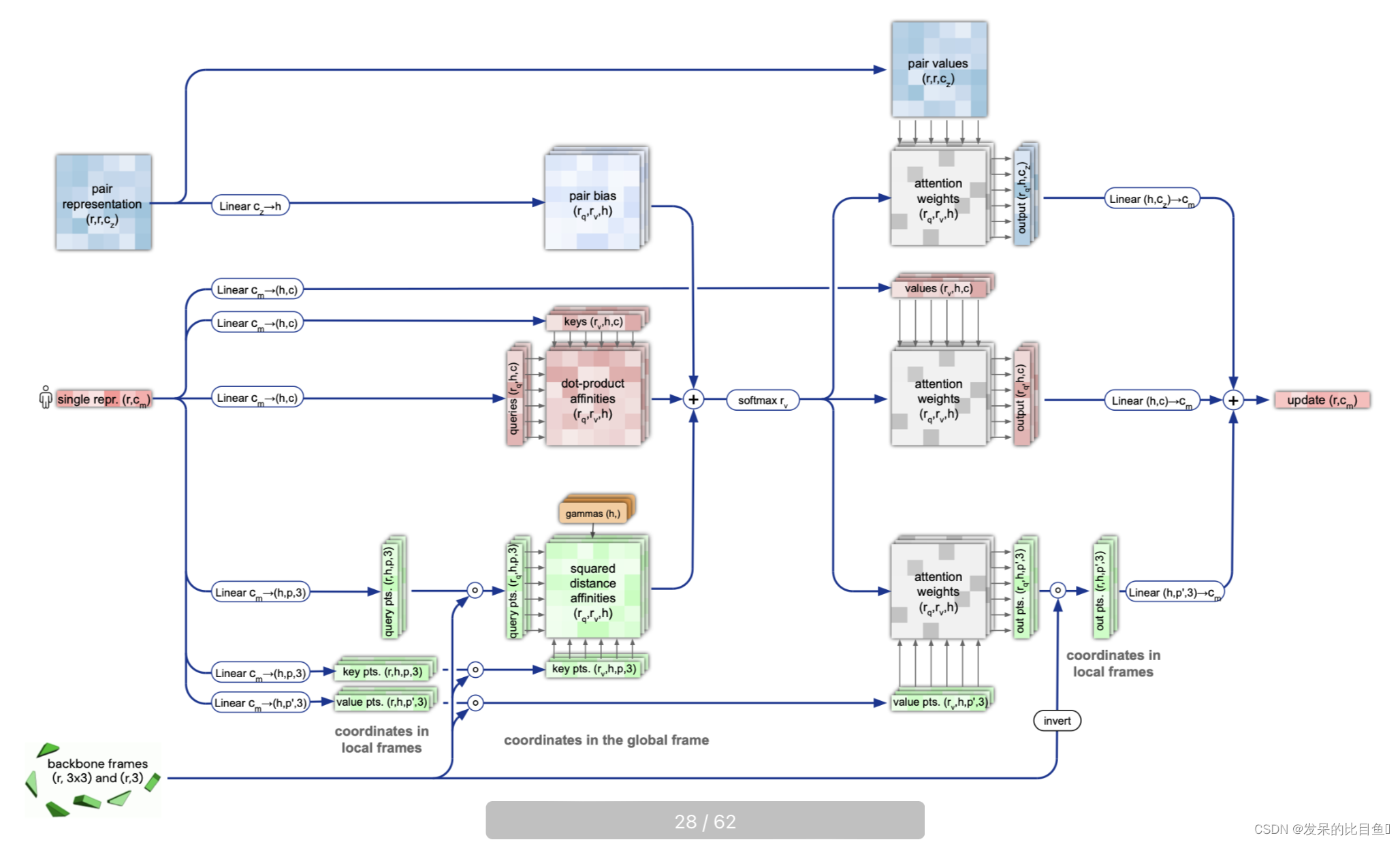

不变点注意(IPA)是一种作用于一组frames的注意力形式,在上述frames的全局欧几里得变换
T
g
l
o
b
a
l
T_{global}
Tglobal下是不变的。我们用纳米表示IPA内的所有坐标;单位的选择会影响注意力亲和力的点分量的比例。
为了定义不同术语的初始权重,我们假设所有的查询和钥匙都来自单位正态分布N(0, 1)的iid,并计算注意力对数的方差。每个标量对 q q q, k k k的贡献是 V a r [ q k ] = 1 Var[qk]=1 Var[qk]=1。每个点对( q ⃗ , k ⃗ \vec{q},\vec{k} q,k)贡献 V a r [ 0.5 ∣ ∣ q ⃗ ∣ ∣ 2 − q ⃗ T k ⃗ ] = 9 / 2 Var[0.5 ||\vec{q}||^2- \vec{q}^T\vec{k}] = 9/2 Var[0.5∣∣q∣∣2−qTk]=9/2。加权因子 w L w_L wL和 w C w_C wC的计算结果是,所有三个项的贡献相等,而且所得方差为1。每头的权重 γ h ∈ R γ^h\in R γh∈R是可学习标量的软加法。
不变性的证明是直截了当的。全局变换在亲和力计算中被抵消,因为矢量的L2-norm在刚性变换下是不变的。
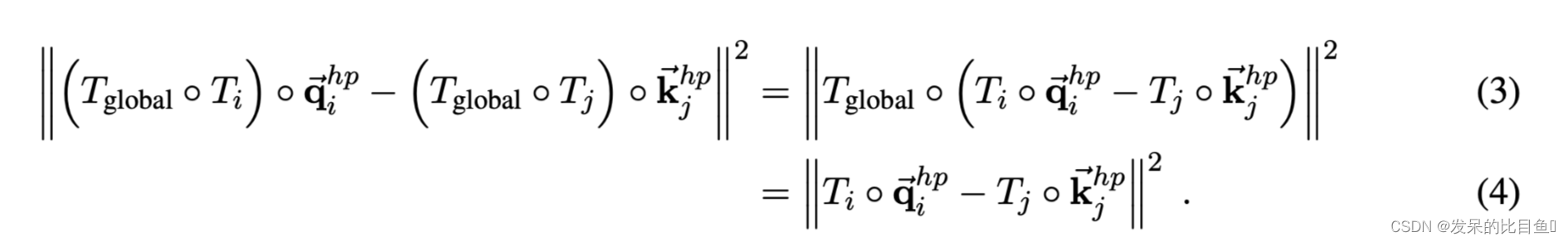
在输出点的计算中,它在映射回本地frame时被抵消了。

相对于全局参考frame的不变性反过来又意味着,在保持嵌入固定的情况下,对所有的残差应用一个共同的刚性运动,将导致局部frame中的相同更新。因此,更新的结构将被相同的共享刚性运动所转化,这表明这一更新规则在刚性运动下是等值的。在这里和其他地方,"刚性运动 "包括适当的旋转和平移,但不包括反射。
骨干网更新
骨干frame的更新是通过预测一个用于旋转的四元数和一个用于平移的矢量创建的。非单位四元数的第一个分量被固定为1。定义欧拉轴的三个分量是由网络预测的。 这个程序保证了有效的归一化四元数,而且有利于小旋转而不是大旋转(四元数(1,0,0,0)是同一旋转)。

计算所有的原子坐标
结构模块预测骨干frame T i T_i Ti和扭转角 α ⃗ i f \vec{\alpha}^f_i αif。然后,通过将扭角应用于相应的氨基酸结构,以理想化的键角和键长构建原子坐标。 我们给每个刚体组附加一个局部frame,这样扭转轴就是X轴,并将每个氨基酸相对于这些frame的理想原子坐标存储在一个表 x ⃗ r , f , a l i t \vec{x}_{r,f,a}^{lit} xr,f,alit中,其中KaTeX parse error: Expected 'EOF', got '}' at position 28: … ARG, ASN, ... }̲表示残基类型, f ∈ S t o r s i o n n a m e s f \in S_{torsion names} f∈Storsionnames表示frame, a a a表示原子名称。 我们进一步预先计算刚性转换,将原子坐标从每个frame转换到层次结构中更高的frame。 例如, T r , ( χ 2 → χ 1 ) l i t T^{lit}_{r,(χ2→χ1)} Tr,(χ2→χ1)lit将氨基酸类型 r r r的原子从 χ 2 χ^2 χ2frame映射到 χ 1 χ1 χ1frame。由于我们只预测重原子,额外的骨架刚性基团ω和φ不包含原子,但相应的frame会导致FAPE损失,从而与真实结构对齐(与所有其他框架一样)。


重命名对称真实原子
一些刚性基团的180◦旋转对称性导致该基团中所有不在旋转轴上的原子在命名上出现模糊。
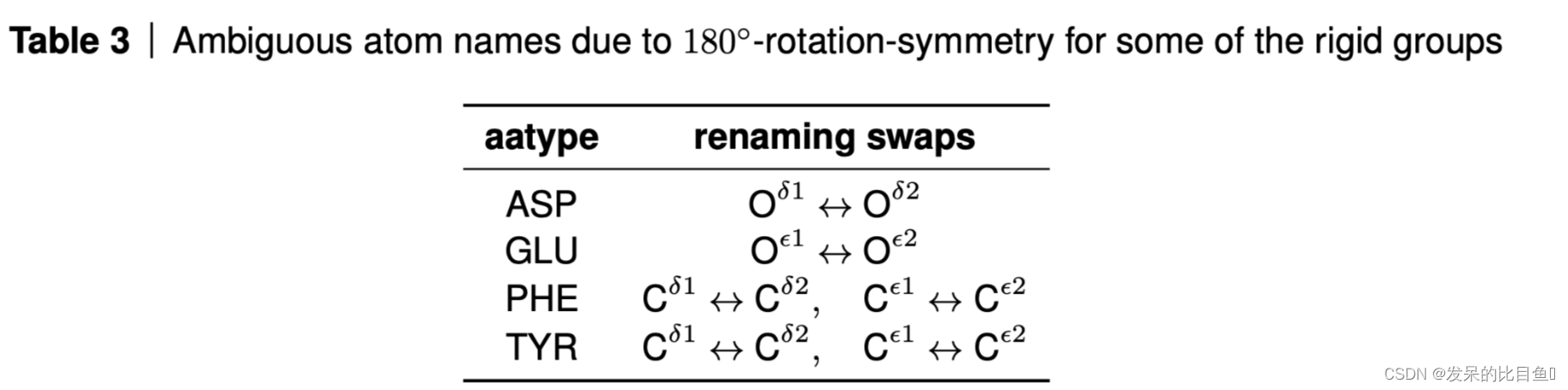
算法26以全局一致的方式通过重新命名基础真理结构来解决命名的模糊性。对于每个残基,它计算原子的lDDT,对照所有非歧义原子的基础真知原子的两种可能命名("真 "和 “替代真”)。
非模棱两可的原子集合
S
n
o
n
−
a
m
b
i
g
u
o
u
s
a
t
o
m
s
S_{non-ambiguous atoms}
Snon−ambiguousatoms是表2的所有元组(residue-type, atom-type)减去表3的模棱两可的原子集合。随后,该算法对模糊的基础事实原子进行重新命名,使其与预测结构最匹配。

Amber 优化
为了解决任何剩余的结构违规和冲突,我们通过迭代约束能量最小化能力优化模型预测。这部分对结果没什么影响,我们省略。。。。