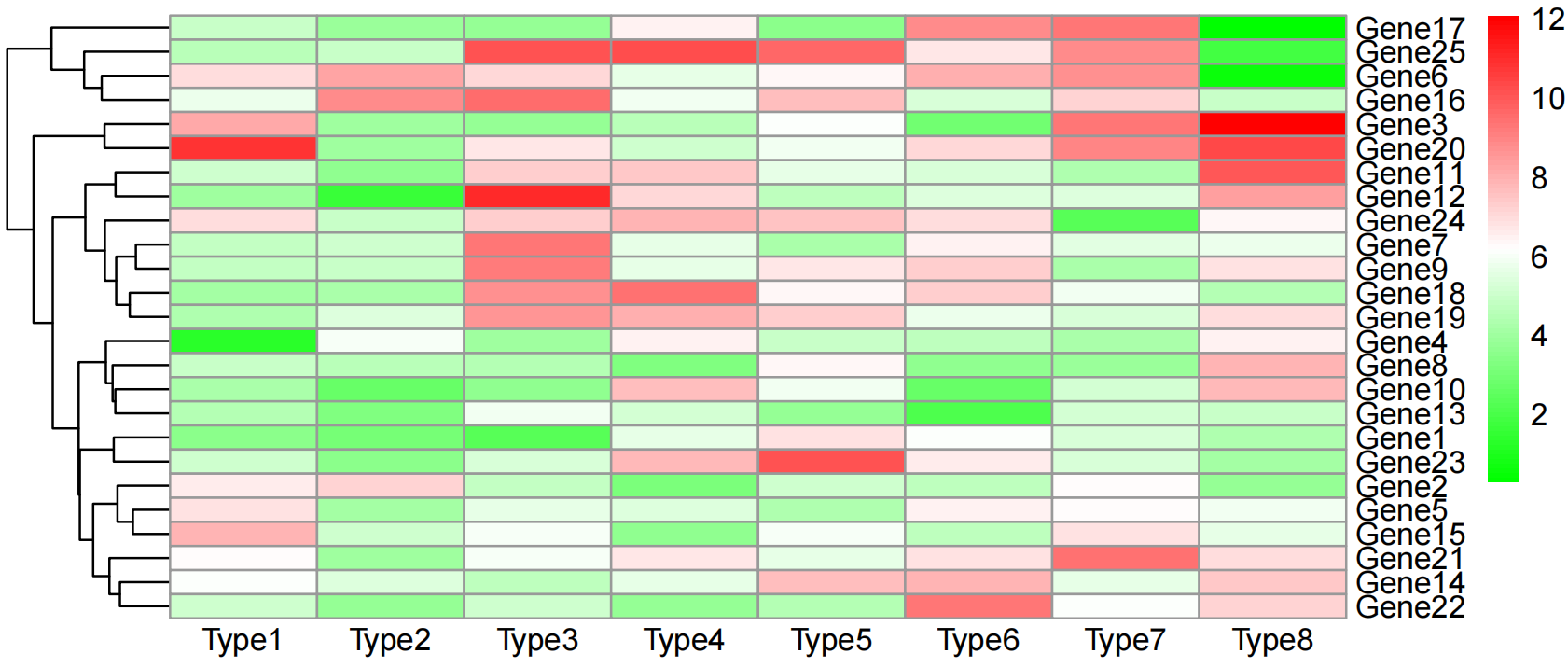
Pheatmap绘制基因表达量热图
论文中展示基因表达量变化通常使用热图,今天分享一个快速绘制不同基因在各处理下表达量变化的方法,使用R语言中pheatmap包,它可以用于可视化数据集中的数值,以便更好地理解数据之间的关系和模式。
创建环境与示例数据
加载R包
library(tibble)
library(tidyverse)
library(pheatmap)
生成随机数据
# 设置随机数种子以确保结果可重复
set.seed(1234)
# 生成随机矩阵
expr_mat <- matrix(rnorm(200, mean = 6, sd = 2), ncol = 8)
# 将矩阵转换为tibble,并设置行名称和列名称
expr_mat <- as_tibble(expr_mat, colnames = paste0("Type", 1:8))
colnames(expr_mat) <- paste0("Type", 1:8)
rownames(expr_mat) <- paste0("Gene",1:25)
# 查看结果
head(expr_mat)
通过上述代码可以生成一个随机的基因表达矩阵,其中每行是一个基因,每列代表一个处理,共有25行8列,展示了25个不同的基因在8个不同处理下表达量的变化情况。数据内容如下:
> head(expr_mat)
# A tibble: 6 × 8
Type1 Type2 Type3 Type4 Type5 Type6 Type7 Type8
<dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 3.59 3.10 2.39 5.69 6.83 6.16 5.25 4.32
2 6.55 7.15 4.84 3.22 5.05 4.74 6.20 3.75
3 8.17 3.95 3.78 4.55 6.13 2.97 9.28 12.1
4 1.31 5.97 3.97 6.52 5.00 4.73 4.25 6.47
5 6.86 4.13 5.68 5.37 4.35 6.45 6.24 5.93
6 7.01 8.20 7.13 5.64 6.33 8.03 8.72 0.536
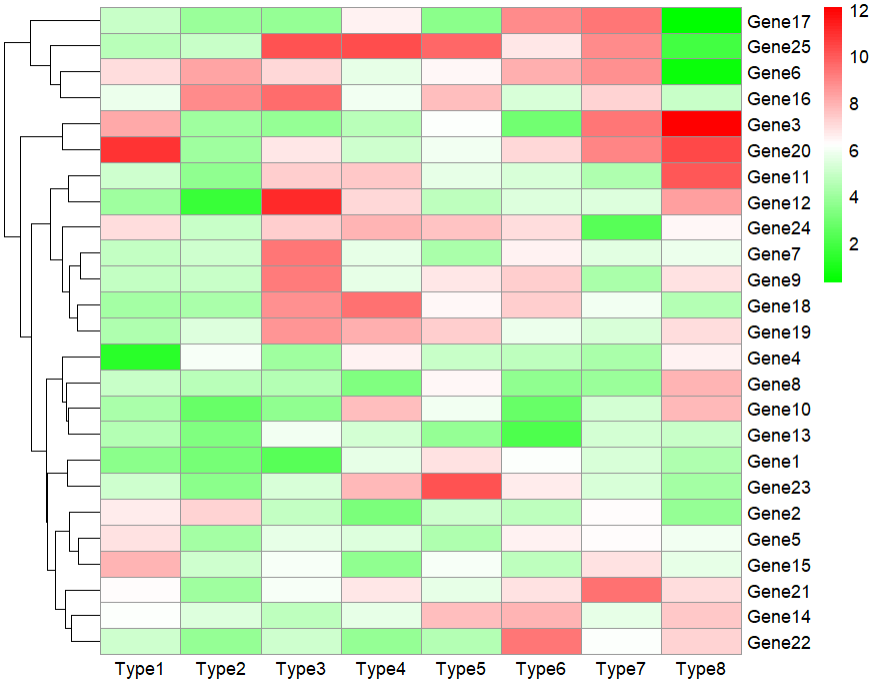
绘制图像
基础版热图
pheatmap(expr_mat)
直接运行pheatmap函数,输入数据矩阵,即可快速生成漂亮的热图,会自动对行和列进行聚类,这也是最简单的热图生成方法。
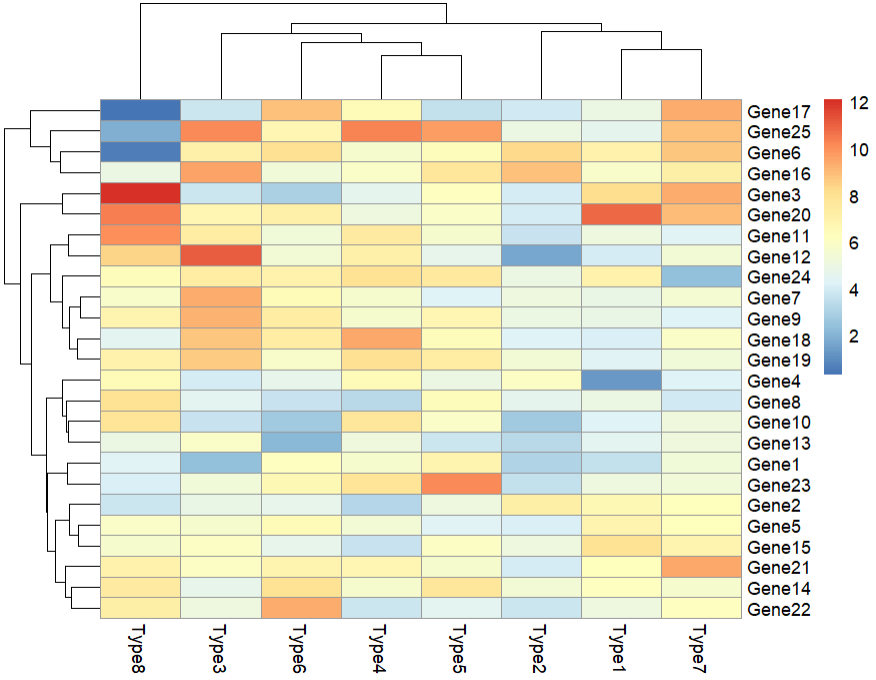
进阶版热图
my_palette <- colorRampPalette(c("white", "yellow","red"))(n = 100)
pheatmap(expr_mat,
cluster_cols = F,
cluster_rows = T,
filename = "GeneExpr.pdf",
color = my_palette,
width = 2.35*3,
height = 1*3,
angle_col = 0)
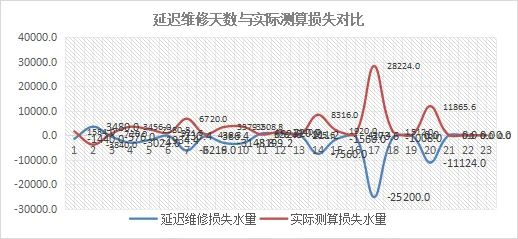
通常绘制基因表达热图的目的是看不同基因和处理之间的变化关系,比如不同时间下基因的表达量变化,因此不需要对处理进行聚类,为了让图中横轴Type按照顺序排列,取消纵轴聚类,可以使用cluster_cols = F参数,另外可以自定义修改配色的方案,使用colorRampPalette可以自定义颜色。
上面的代码会在当前工作目录下生成一个pdf图片文件,如果想让pheatmap绘制的图形直接显示在Rstudio的右下角plot窗口,删除filename = "GeneExpr.pdf"参数即可。
pheatmap 函数可以根据数据集的值自动为每个单元格分配颜色,并且可以对行和列进行聚类以更好地显示数据之间的关系。此外,pheatmap 还支持自定义颜色映射,使用户可以将数据映射到自定义颜色范围中。
以下是 pheatmap 函数的一些常见参数:
data:要绘制的数据集。scale:是否对数据进行标准化。默认为 “row”,表示对每一行进行标准化。也可以设置为 “column” 或 “none”。color:颜色映射。可以使用内置的颜色映射,也可以使用colorRampPalette函数创建自定义颜色映射。cluster_rows和cluster_cols:是否对行和列进行聚类。默认为 TRUE。show_rownames和show_colnames:是否显示行名和列名。默认为 TRUE。fontsize_row和fontsize_col:行名和列名的字体大小。默认为 12。
本文由mdnice多平台发布