继续我们的跟着NC学系列,前面分享的是关于16S扩增子测序和宏基因组数据分析的。考虑到我们有许多小伙伴是做人类基因组方面的,这次分享一篇癌症早筛方面的,血液DELFI全基因组片段化丰度谱检测的分析框架。题目是:Detection and characterization of lung cancer using cell-free DNA fragmentomes。

文章虽然不是特别新,发表于2021年, 可代码和数据公开地相当彻底,甚至包括建模代码和模型,适合我们学习。文章主要内容是研究者对LUCAS发现队列(365位受试者)血液样本cfDNA进行低深度全基因组测序,计算获得全基因组片段化丰度谱,并且基于此利用机器学习构建了DELFI肺癌诊断模型并且进行交叉验证。将独立验证队列(431位受试者)用于评估肺癌诊断模型的表现,证明了DELFI在早期肺癌的诊断作用。研究思路如下图所示:
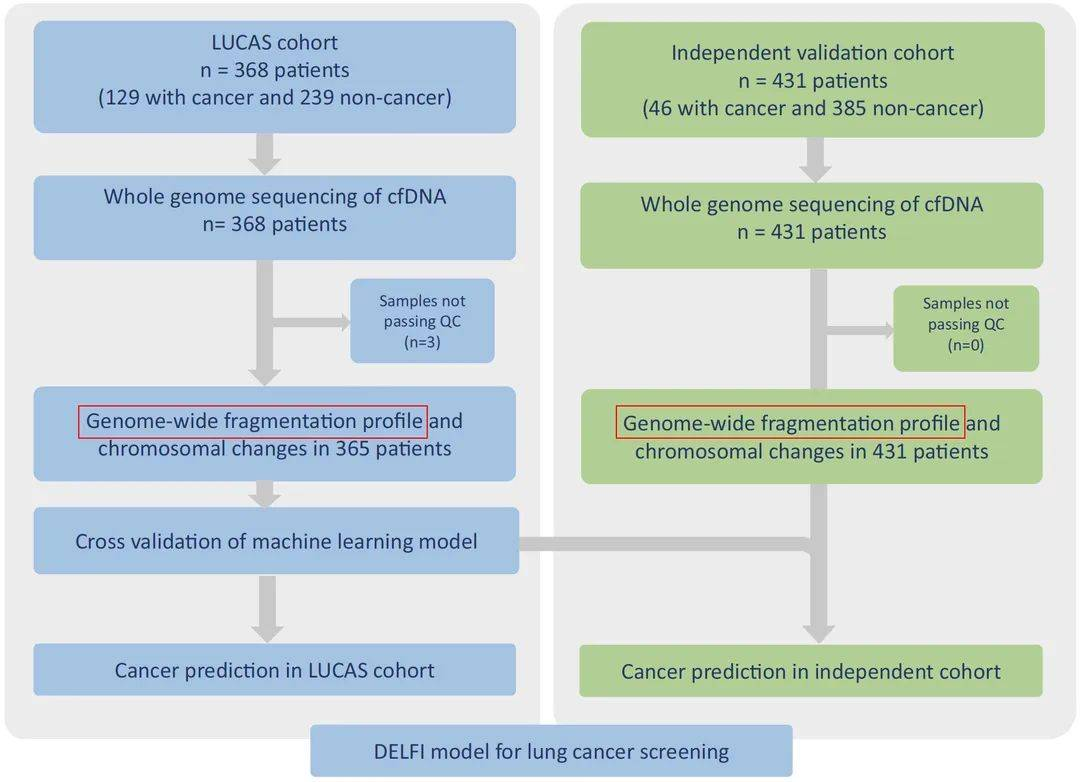
关于文章内容的解读方面,已经有很多啦,这里不再重复,大家可以搜索,比如这个公众号的推文,算法原理方面解读地也比较清楚啦,这里重点分享一下分析代码方面的。
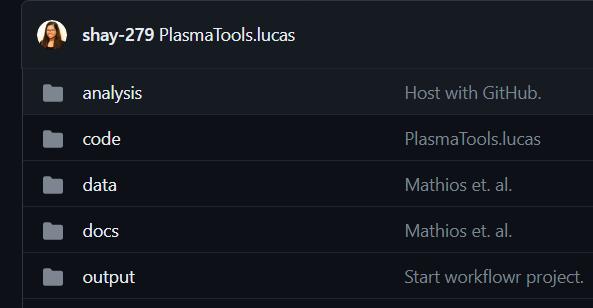
repo总体结构
此workflowr中有4个文件夹。
-
(1) analysis -包含生成论文每个图形所需的代码。
-
(2) code -这包含rpcr和rLucas(分析中广泛使用的两个包)、一些具有必要功能的单独r脚本,以及与模型创建有关的4个文件夹。
对3个队列的模型进行了训练和评估:a.LUCAS队列(158例非癌症,129例癌症)。b.排除既往癌症的LUCAS队列。c.LUCAS队列不包括以前的癌症,仅限于50-80岁的吸烟者。
MODEL_CODE具有训练每个队列上的模型以及执行来自第一个队列的模型的外部验证所需的代码。Models_c1、Models_c2和Models_c3包含训练好的模型。对于前两个队列,培训了两个模型架构,对于第三个队列,只训练了一个。
-
(3) data -这包含用于训练模型和生成图形的原始数据。
-
(4) docs -包含分析中的markdown和html,以及生成的图形。
这个存储库可以在Github上获得,可以作为一个workflowr运行,以生成一个链接了所有代码和图形的网页。
代码概览
前处理
代码主要集中在code/preprocessing文件夹。
首先,precess.sh脚本调用以下脚本,这些脚本执行以下操作:
fastp.sh--将FASTQ使用fastp质控。align.sh--bowtie2和samtools生成bam文件。post_alignment.sh--sambamba和samtools生成bed文件。bed_to_granges.sh--将前面步骤生成的bed文件转换为R中的Granges。gc_count ts.sh--为每个GC层的片段计数创建一个表。用于在片段级进行GC校正。bin_right ted.sh--为每个样本创建bin级别数据。
然后,分别运行getZcore res.sh、getCoverage.sh以生成z-Score和Coverage特性。
最后,FeatureMatrix.sh生成可用于后续建模的最终特征矩阵。
create-training-set.r和create-testing-set.r和生成完整的特征矩阵,包括临床数据,用于训练和验证本文使用的模型。
必须从GitHub安装以下库:
看起来是不同的测序仪也需要相应的校正,可见研究还是很严谨的。
用于目标GC分布的PlamaToolsNovaseq.hg19和用于noveseq参考样品的bins(未在论文中使用)。
https://github.com/cancer-genomics/PlasmaToolsNovaseq.hg19。
用于目标GC分布的PlamaToolsHiseq.hg19和用于HiSeq参考样品的bins(使用)。
https://github.com/cancer-genomics/PlasmaToolsHiseq.hg19
以precess.sh和fastp.sh为例来欣赏下代码,很规范和整洁,qsub任务提交,注释清晰,值得我们好好学习。想要复现文章的结果,基本上只需要修改下软件和参考文件的路径即可。
首先来看precess.sh:
#!/bin/bash
#$ -cwd
#$ -j y
#$ -l mem_free=1G
#$ -l h_vmem=1G
# Job resource option: max runtime
#$ -l h_rt=96:00:00
qsub fastp.sh
qsub -hold_jid_ad fastp.sh align.sh
qsub -hold_jid_ad align.sh post_alignment.sh
qsub -hold_jid_ad post_alignment.sh,align.sh bed_to_granges.sh
qsub -hold_jid_ad post_alignment.sh,bed_to_granges.sh,align.sh gc_counts.sh
qsub -hold_jid post_alignment.sh,bed_to_granges.sh,gc_counts.sh,align.sh bin_corrected.sh
再来看下fasp.sh:
#!/bin/bash
#$ -cwd
#$ -j y
#$ -l h_fsize=100G
#$ -l mem_free=10G
#$ -l h_vmem=10G
#$ -l h_rt=96:00:00
## #$ -pe local 8
#$ -t 1-41
module load bowtie2 # 使用了module,感兴趣的同学可以搜索相关教程进行学习
module load samtools
PATH="/users/scristia/.linuxbrew/bin:$PATH"
FASTP=/users/scristia/fastp
CORES=1 ##CORES=8
umask g+w
# Inputs,文件夹设置很洁癖
fqdir="../fastq"
outdir="../bam"
beddir="../bed"
scratchdir="tmp"
outdirfastp=$outdir/fastp
# Main
mkdir -p $outdir $outdir/dup_metrics $outdir/fastp $beddir $scratchdir
samplepath=$(find $fqdir -maxdepth 1 -name "*.fastq.gz" | \
sed 's/.\{12\}$//' | \
sort -u | \
head -n $SGE_TASK_ID | \
tail -n 1)
sample=$(basename $samplepath | awk '{ gsub("_WGS", "") ; print $0 }')
echo $sample
read1fq=$samplepath\_R1.fastq.gz
read2fq=$samplepath\_R2.fastq.gz
# Trimming with fastp
echo Trimming $sample with fastp
read1fqpaired=$scratchdir/$sample.paired.R1.fastq.gz
read2fqpaired=$scratchdir/$sample.paired.R2.fastq.gz
if [ -f $read1fqpaired ]; then
echo Sample $sample has been processed. Exiting.
exit 0
fi
json=$outdirfastp/$sample.json
html=$outdirfastp/$sample.html
$FASTP -i $read1fq -I $read2fq -o $read1fqpaired -O $read2fqpaired \
-Q -z 1 --detect_adapter_for_pe -j $json -h $html -w $CORES
建模
也是非常详尽的代码,一步一步完整公开,这里就不再列出了,感兴趣的同学可以直接阅读原文查看repo的code/model_code/文件夹。当然,作者2019年也发表过一篇NC,代码公开也很完整,这里也放在这(文末的参考4和5),供大家参考对比。
分享就先到这里啦,学起来呀!虽然人工智能时代强势来临,这对于我们来说可能是更多地学习,用好AI,如果不想躺平的话,加油各位!
附:
1.workflowr简介
R中有组织的 + 可重现 + 可共享的数据科学框架,Workflowr结合了编程(knitr和rmarkdown)和版本控制(通过git2r的Git)来生成一个包含时间戳记,版本控制和文档化的结果的网页。任何R用户都可以快速轻松地使用它。其设计的初衷是助研究人员以促进有效的进行项目管理,可重复性的分析,同时进行协作和对结果进行共享。
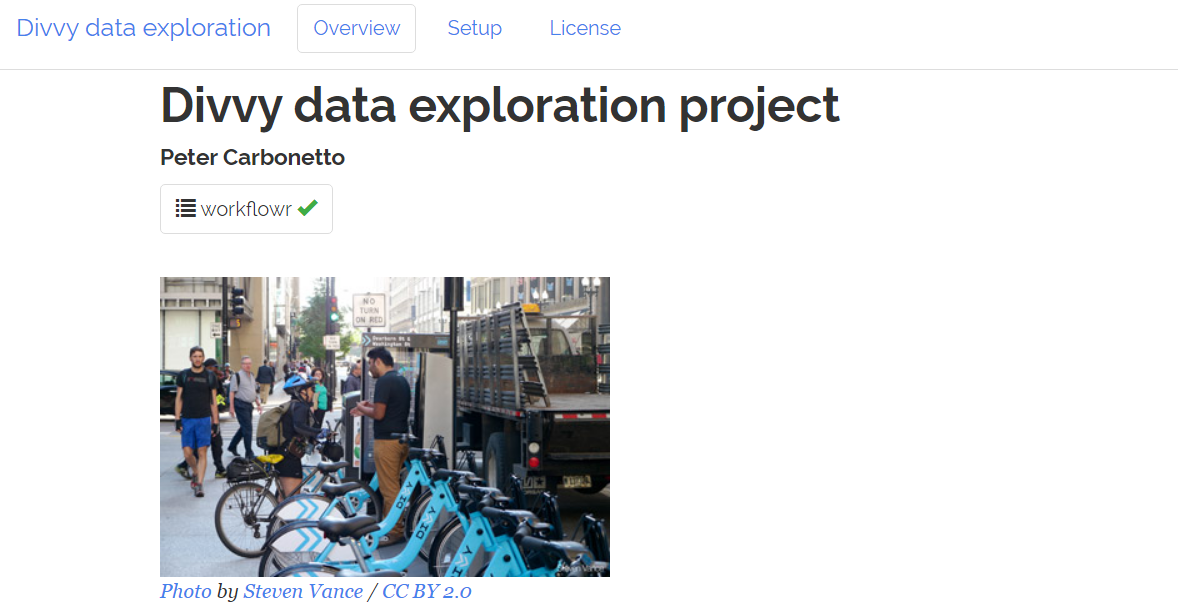
2. 一个缺少文件的处理
在学习使用的过程中,发现code/preprocessing/01-bed-to-granges.r中缺少cytosine_ref.rds这么个文件,如果对基因组不太熟悉可能不太好解决,有帅哥提出了这个issue。作者也贴心地给出了解决方案和原因(文件体积太大,有3G), 附在这里:
# repo里多数包是用bioconductor安装的
library(GenomicRanges)
library(Biostrings)
library(BSgenome.Hsapiens.UCSC.hg19)
library(Homo.sapiens)
cytosines <- vmatchPattern("C", Hsapiens)
saveRDS(cytosines, 'cytosine_ref.rds')
参考资料:
[1] Mathios, D. et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat Commun 12, 5060, doi:10.1038/s41467-021-24994-w (2021).
[2] https://www.sohu.com/a/575968290_100148274?qq-pf-to=pcqq.group
[3] https://github.com/cancer-genomics/reproduce_lucas_wflow
[4] Ulz, P. et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat Commun 10, 4666, doi:10.1038/s41467-019-12714-4 (2019).
[5] https://github.com/cancer-genomics/delfi_scripts




![[组合数学]母函数与递推关系](https://img-blog.csdnimg.cn/ae5f58496c0e46668a549328fe1bbf61.png)