AMBER分子动力学模拟之结果分析(构象分析)-- HIV蛋白酶-抑制剂复合物(6)
RMSD RMSF b-facto计算
RMSD
RMSD measures the deviation of a target set of coordinates (i.e. a structure) to a reference set of coordinates, with
R
M
S
D
=
0.0
\mathrm{RMSD}=0.0
RMSD=0.0 indicating a perfect overlap. RMSD is defined as:
R
M
S
D
=
∑
i
=
0
N
[
m
i
∗
(
X
i
−
Y
i
)
2
]
M
R M S D=\sqrt{\frac{\sum_{i=0}^N\left[m_i *\left(X_i-Y_i\right)^2\right]}{M}}
RMSD=M∑i=0N[mi∗(Xi−Yi)2]
B-Factoryl温度因子|德拜-沃勒因子
作用:B因子体现晶体中原子电子密度的“模糊度”(diffusion),这个“模糊度”实际上反映了蛋白质分子在晶体中的构象状态.B因子越高,“模糊度”越大,相应部位的构象就越不稳定。在晶体学数据中,B因子一般是以原子为单位给出的,我们可以换算成相应残基的B因子,从而分析残基的构象稳定性。
RMSF
RMSF与B因子换算公式:
R
M
S
F
2
=
3
B
8
π
2
R M S F^2=\frac{3 B}{8 \pi^2}
RMSF2=8π23B
tleap.in
parm ../top/com.top
trajin ../md/md1.crd 1 last 1
trajin ../md/md2.crd 1 last 1
reference top/com.pdb
center :1-198 mass
image center familiar
rms reference @CA
##rmsd
rms reference mass out rms_pro.dat :1-198@CA,C,N time 0.002
rms reference mass out rms_lig.dat :199 time 0.002
##b-factor
atomicfluct out bfactor_residue.dat :1-198@CA,C,N byres bfactor
atomicfluct out bfactor_allatom.dat :1-198@CA,C,N byatom bfactor
## rmsf
atomicfluct out rmsf_residue.dat :1-198@CA,C,N byres
atomicfluct out rmsf_allatom.dat :1-198@CA,C,N byatom
执行
cpptraj -i tleap.in

可视化
xmgrace rms_pro.dat
计算rms2d
tleap.in
parm ../top/com.top
trajin ../md/md2.crd
rms2d mass :1-198@CA,C,N out 2drms_pro.gnu time 0.0002
输出的gnuplot格式的图片,可以使用gnuplot打开
距离 角度计算
tleap.in
parm ../top/com.top
# trajin ../md/md1.crd
trajin ../md/md2.crd
## MOL_199@O5 ILE_149@H ILE_149@N
distance dist1 :149@N :199@O5 out dis.dat time 0.0002
## MOL_199@O5 ILE_149@H ILE_149@N
angle angle1 :199@O5 :149@H :149@N out angle.dat time 0.0002
执行
cpptraj -i tleap.in

SASA计算
可及表面积(ASA,accessible surface area)或溶剂可及表面积(SASA,solvent-accessible surface area)是溶剂可接触的生物分子表面积。ASA的测量通常以平方埃为单位进行描述(分子生物学中的标准测量单位)。ASA由Lee&Richards于1971年首次描述,有时被称为Lee-Richards分子表面。ASA通常使用Shrake&Rupley在1973年开发的“滚球“算法来计算。该算法使用特定半径的(溶剂的)球体来“探测”分子的表面。
与范德华表面相比,溶剂可及表面如下左图。由原子半径给出的范德华表面以红色显示。可触及的表面用虚线绘制,并且是通过沿球体范德华表面滚动跟踪球体的中心(蓝色)而创建的。请注意,此处描绘的探针半径的比例尺小于典型的1.4A。溶剂分子在蛋白质的范德华表面上滚动时的中心,如下右图所示(Gromiha和Ahmad,2005)。通常,假定水球是半径为1.4A的溶剂分子。溶质分子由分配给每个原子的适当范德华半径的一组互锁球表示(interlocking spheres),溶剂分子沿范德华表面的外壳在方便剖切的平面上滚动。因此,半径为r的原子的ASA是半径为R=r+rsolv的球体表面上的区域,在该区域的每个点上,可以使溶剂分子的中心与该原子接触,而不会穿透任何其他原子溶质分子。

parm ../top/com.top
# trajin ../md/md1.crd
trajin ../md/md2.crd
surf out surf.dat
surf :1-10 out surf_10.dat
surf :11-20 out surf_20.dat
surf :1-20 out surf_all.dat
执行
cpptraj -i tleap.in

氢键作用计算
# Hydrogen bond analysis with cpptraj
# Load topology and trajectory
#trajin ../md1.crd
#trajin ../md2.crd
#trajin ../md3.crd
#trajin ../md4.crd
trajin ../md/md3.crd
# hbond of protein and ligand
hbond angle 120.000 dist 3.500 donormask: 1-306 acceptormask:307 out nhb.dat avgout avghb.dat
hbond angle 120.000 dist 3.500 donormask: 224 acceptormask:1-223 out nhb.dat avgout avghb.dat
#hbond of protein and bridging water
#hbond angle 120.000 dist 3.500 donormask:1-223 acceptormask:232 out nhb.dat avgout avghb.dat
#hbond angle 120.000 dist 3.500 donormask:232 acceptormask:1-223 out nhb.dat avgout avghb.dat
#hbond of liqand and bridqing water
#hbond angle 120.000 dist 3.500 donormask:224 acceptormask:232 out nhb.dat avgout avghb.dat
#hbond angle 120.000 dist 3.500 donormask:232 acceptormask:224 out nhb.dat avgout avghb.dat

cluster 计算
cluster这个词,可以翻译成团簇,也可以翻译成聚类.虽然数学上来说实质相同,当具体使用的时候还是有些不同的,适当加以区分可以更明确,避免一些模糊之处.
在MD的语境下,我们说进行团簇分析,一般指的是,给定一个构型,其中包含多个原子,我们按一定的规则(常用的是距离)将这些原子归属到不同的聚集体,也就是团簇中,这样可以将体系划分为一些团簇,对每个团簇进行分析,获取一些信息.

聚类的过程,以分子对接为例子。对接后各个对接构型按打分从高到低排序。
- 第一个构型是第一类,第一类构型数量是一。
- 第二个构型与第一类中打分最高的构型比较一下rmsd。若rmsd小于2,则第二个构型属于第一类,该类构型数量加一;若rmsd大于2,则新建一个类,第二个构型属于新建的一类,该类的构型数量是一。
- 第n个构型依次与上面各个类中打分最高的构型比较rmsd,若与第m类rmsd小于2,则属于第m类,第m类构型数量加一,结束计算与m+1类的rmsd;若与所有类的rmsd都大于2,则新建一个类,第n个构型属于新建的一类,该类的构型数量是一。
tleap.in
parm ../top/com.top
# trajin ../md/md1.crd
trajin ../md/md2.crd
cluster hieragglo epsilon 1.0 clusters 5 averagelinkage rms mass @CA,C,N out frame2cluster.dat summary summary.dat repout Cluster repfmt pdb
基于量子计算, 半经验的, qm-mmpbsa
tleap.in
&general
startframe=1,
endframe=1000,
interval=5,
verbose=2,
keep_files=2,
/
&gb
igb=1,
ifqnt=1,
qmcharge_com=1,
qm_residues='27,28,30,32,47,81,84,124,129,148,149,180,183,199',
qm_theory='PM6-D',
qmcharge_lig=0,
qmcharge_rec=1,
/
MMPBSA.py -O -i tleap.in -o qm_mmgbsa.dat -cp ../top/com.top -rp ../top/pro.top -lp ../top/lig.top -y ../md/md2.crd > qm_mmgbsa.log
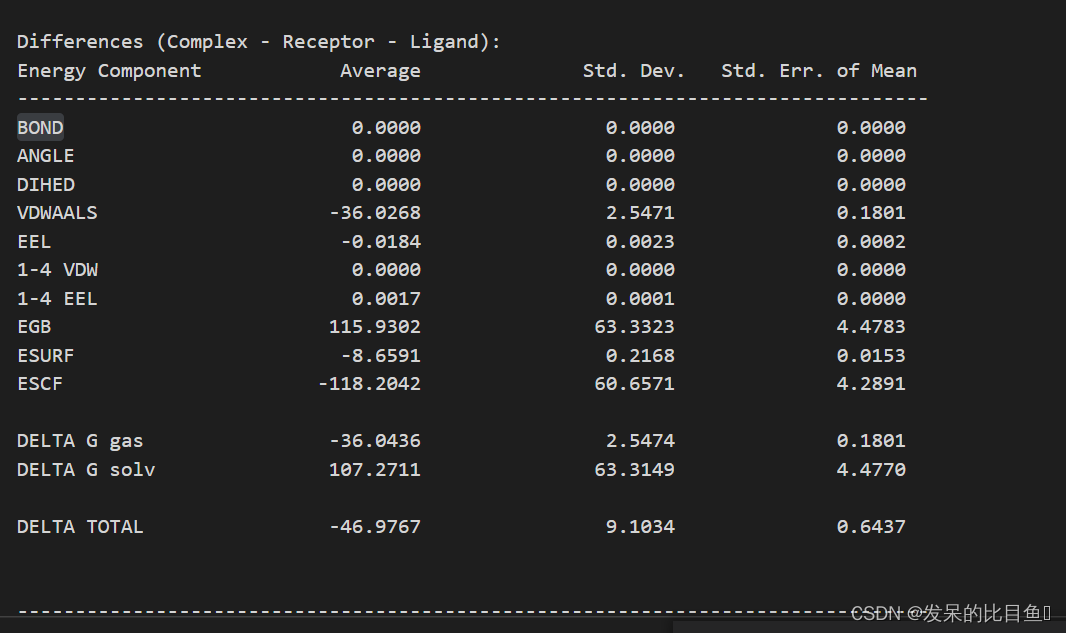
找残基id 找2.5埃的残基
parm ../top/com.top
trajin ../md/md2.crd 1 last 1
reference ../top/com.pdb
center :1-198 mass
image center familiar
rms reference @CA
### rmsf
atomicfluct out rmsf_residue.dat :199<@2.5 byres
可视化
xmgrace *.dat



![[离散数学]谓词逻辑与推理演算](https://img-blog.csdnimg.cn/9d21fc8f16c74ad1b6d936e243eb5c0a.png)