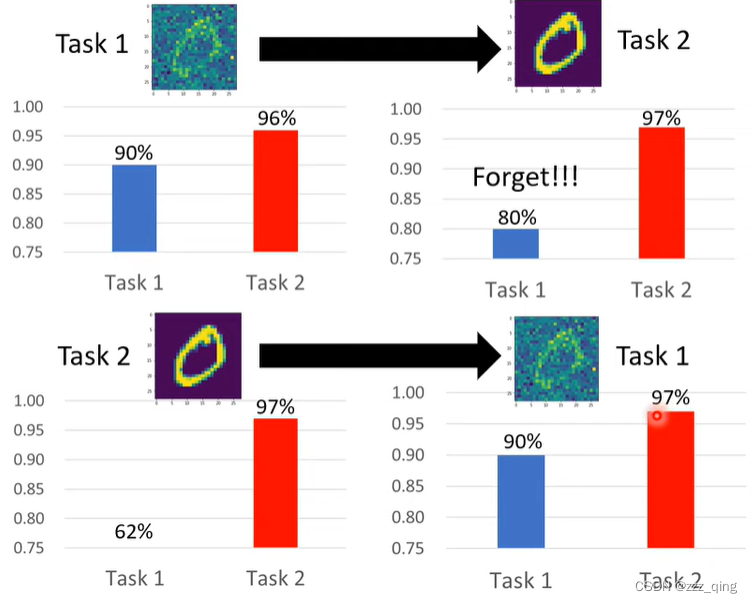
编译 | 于洲
今天我们介绍来自复旦大学的Lihao Wang以及其他来自字节跳动AI实验室与清华大学AI产业研究院的成员发布在NeurIPS 2022会议上的工作,该文章介绍了一种新方法——正则化分子构象场(RMCF),用于从化学结构中预测最有利的三维构象。文章强调了小分子在自然条件下可能出现的复杂动力学,这可能导致高维度的势能面(PES),从而使得从PES中获得多样的样本变得困难。为此,作者们提出了RMCF,该方法利用图神经网络计算分子结构的表示,并通过正则化技术进行优化。文章还详细介绍了RMCF的计算过程,并提供了开源代码。最后,作者提到了RMCF的局限性。

背景介绍
从分子图中预测有机分子的积极三维构象在计算机辅助药物发现研究中起着重要作用。然而,有效地探索高维构象空间以识别(元)稳定构象绝不是微不足道的。在这项工作中,我们介绍了RMCF,一种新的框架,通过从正则化分子构象场采样来生成多样化的低能分子构象。我们开发了一种数据驱动的分子分割算法,将每个分子自动划分为几个结构构件,以降低建模自由度。然后,我们利用马尔可夫随机场来学习碎片构型和碎片间二面角的联合概率分布,这使我们能够从构象空间的不同低能区域进行采样。
本文的创新与贡献:
本文提出了一种正则化分子构象场的方法,能够从低维输入特征中预测有机分子的三维构象。相较于传统的手工特征提取方法,该方法不需要专业领域知识和人工干预,也能够处理高维度和非线性的输入数据。
本文还提出了一种基于双层编程的构象生成模型,并通过对比实验验证了其在构象生成领域的优越性能。
本文提出的模型可为药物分子设计等领域提供更加准确和高效的工具和方法。
方法介绍
本文提出了一种正则化分子构象场(Regularized Molecular Conformation Fields,RMCF)模型,用于预测有机分子的三维构象。具体来说,RMCF 模型主要由三个步骤组成:
从分子图构建正则化的分子构象空间。在构建分子图方面,采用了分子指纹编码(BRICS)算法,同时对环和侧链进行了切割,防止可能的组合爆炸。具体而言,RMCF 模型通过建立一个分子构象分布,在正则化分子构象空间上进行采样,从而获得多样性的构象样本。

图1:乙烷分子的势能示意图。上图显示了三个简并重叠构象的纽曼投影,下图显示了两个能量有利的交错构象。H-C-C-H二面角足以描述势能的变化
预测分子的二面角和片段构象。在预测分子的二面角和片段构象时,RMCF 模型使用了分层嵌套网络(GNN),将分子图的节点和边分别进行训练。最后,根据预测结果,将分子构建出来。
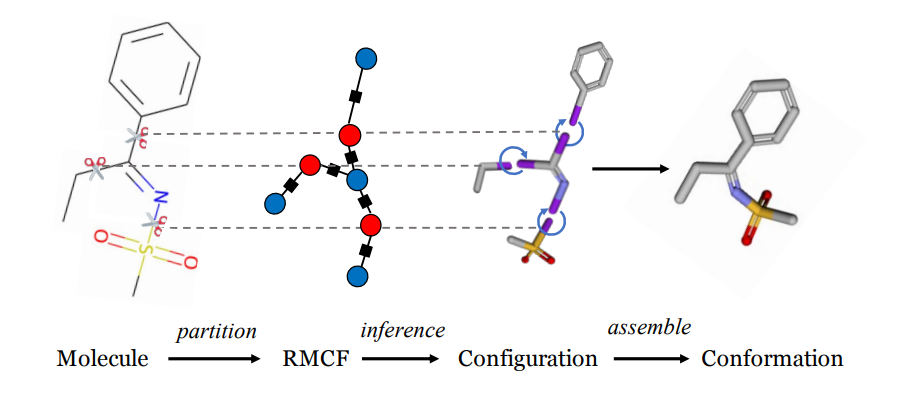
图2:RMCF的工作流程从二维分子图开始,我们将分子划分为片段内自由度最小的片段。蓝色和红色圆圈分别表示碎片和二面角构型,而黑色方块表示相邻构型之间的相互作用。然后,我们使用MRF来模拟碎片和二面体构型的联合概率分布。最后一步是根据预测的二面角和片段构象组装预测的分子构象。
根据预测结果生成分子构象。
实验介绍
数据集:本文使用了两个数据集,一个是QM9,另一个是GEOM-Drugs。其中QM9数据集包含133,885个分子,每个分子都包含了基本的量子力学信息,如化学成分,原子坐标,电子能量等。而GEOM-Drugs数据集则包含了一系列高质量的有机分子构象。实验中,我们采用了GeoDiff论文中的测试集,并按照9:1的比例划分训练集和验证集。最终数据集中,训练集包含271,539个分子,验证集包含30,171个分子,测试集包含1,034个分子。在对GEOM-Drugs数据集进行分子分割后,我们获得了9,081种二维碎片和30,408种三维碎片。对于二面角角度的离散化,我们将360度的区间均匀划分为72个区间。实验采用了信息传递神经网络(Message-Passing Neural Network,MPNN)作为实现图神经网络的框架。
实验过程:本文实验过程分为三个主要步骤。第一步是构造分子图,采用了最少自由度原则,即对于分子的每个部分选择一个旋转角度来代替其所有自由度,并且使用BRICS算法构造分子的化学结构。第二步是预测分子的二面角度,我们采用了一个基于图神经网络和循环神经网络的模型来预测分子的二面角度值。第三步是装配预测的分子构象,根据预测的二面角度和片段构象来组装原子坐标。我们在多个基准数据集上对我们的模型进行了评估,并且取得了竞争性的结果。我们公开发布了我们的代码和模型以及在我们数据集上的预处理结果。
实验结果:本文的实验结果表明,所提出的正则化分子构象场模型(Regularized Molecular Conformation Fields)能够在有限的样本量下,实现高效、准确地预测有机分子的三维构象。具体来说,在多个数据集上进行的实验表明,该模型在预测分子构象时能够显著优于多个现有模型,且其结果具有较好的可解释性。此外,作者还研究了模型表现的上限和下限,并发现使用分子片段作为构象生成的基本单元,极大地简化了模型,避免了需要生成大量无用的变量。总之,该实验结果验证了所提出模型的可行性和有效性。
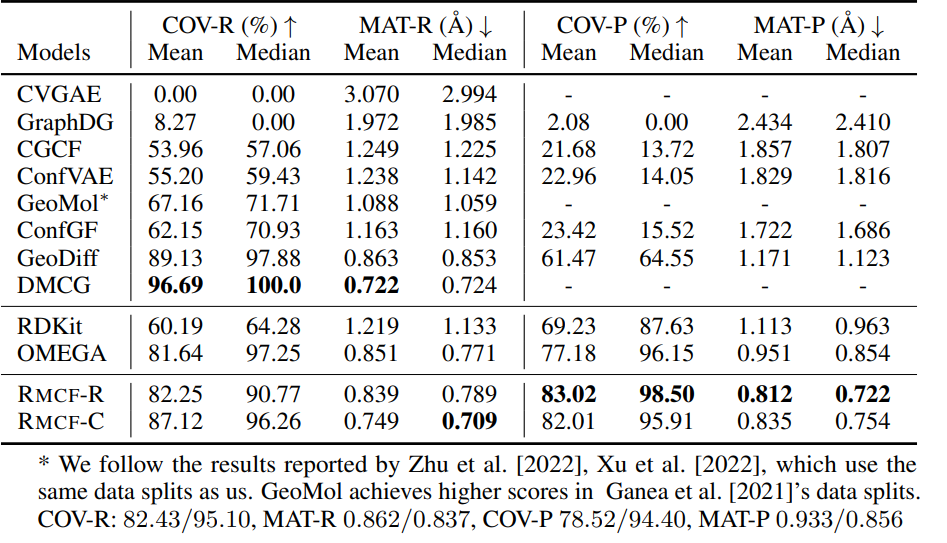
表1:未进行FF优化的GEOM - Drugs数据集的结果
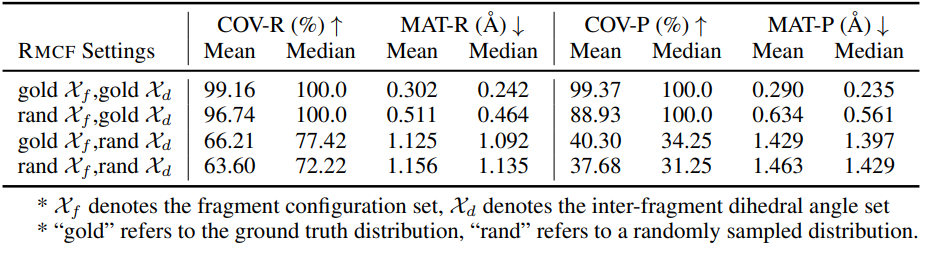
表2:GEOM - Drugs数据集上RMCF性能的经验上界和下界
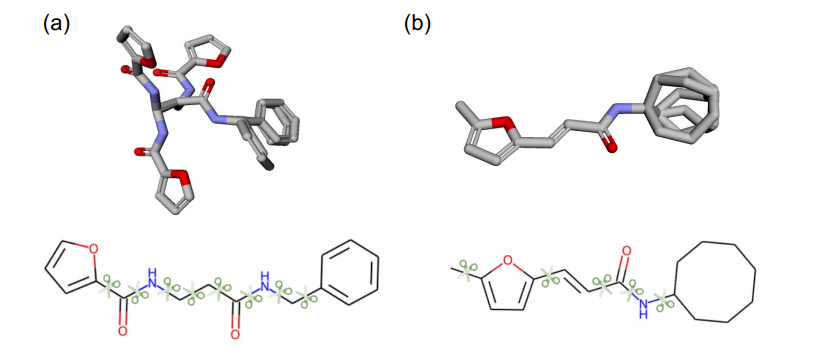
图3:前三个生成的两个例子分子的构象。上面的面板显示了3D原子排列,其中非刚性片段对齐以帮助可视化。下方面板显示了每个分子的分割位置,如剪刀所示。
结论
我们介绍了RMCF,一种新的三维分子构象生成框架。我们的模型是物理驱动的,其中心思想是有效地模拟在减少的构象空间中控制动力学模式的联合概率分布,以实现能量上有利的构象生成。实验结果表明,RMCF在GEOM - Drugs数据集上优于最先进的模型,可以预测位于相应分子势能表面不同局部极小值的不同构象集。我们的方法可以自然地扩展到更大的生物分子系统,例如蛋白质,其构象预测是生物研究界的一个重要主题。
参考资料
Wang, Lihao, Yi Zhou, Yiqun Wang, Xiaoqing Zheng, Xuanjing Huang, and Hao Zhou. "Regularized Molecular Conformation Fields." Advances in Neural Information Processing Systems 35 (2022): 18929-18941.
代码
https://github.com/leowang1217/RMCF