翻译自:AMP - EDA + Models
1.数据集观察
加载四个excel文件
import pandas as pd
train_clinical_data = pd.read_csv('input/train_clinical_data.csv')
train_peptides = pd.read_csv('input/train_peptides.csv')
train_protiens = pd.read_csv('input/train_proteins.csv')
supplemental_clinical_data = pd.read_csv('input/supplemental_clinical_data.csv')
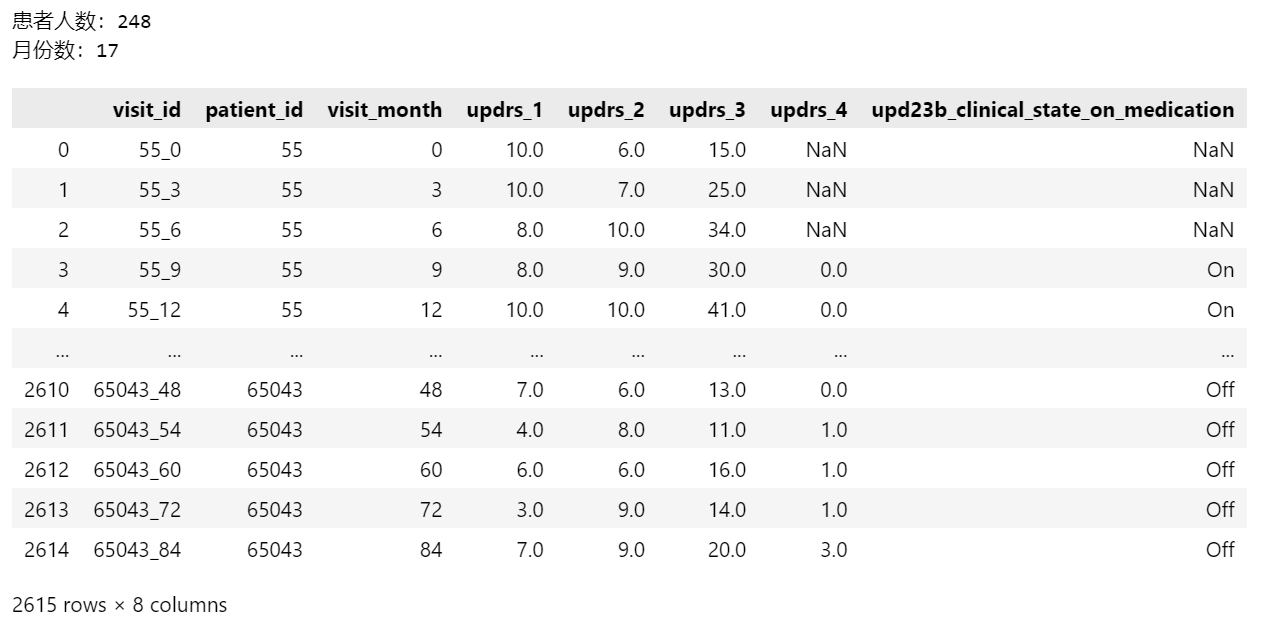
1.1.含脑脊液样本的临床记录数据 来自train_clinical_data.csv
print(f'患者人数:{train_clinical_data["patient_id"].nunique()}')
print(f'月份数:{train_clinical_data["visit_month"].nunique()}')
train_clinical_data
# visit_id 访问的id
# patient_id 患者的id
# updrs_[1-4] 患者评分
# upd23b患者在updors期间是否服用..,主要影响第三部分分数
1.2.肽水平的质谱数据 来自train_peptides.csv
print(f'患者人数:{train_peptides["patient_id"].nunique()}')
print(f'月份数:{train_peptides["visit_month"].nunique()}')
print(f'蛋白质数目:{train_peptides["UniProt"].nunique()}')
print(f'氨基酸序列数:{train_peptides["Peptide"].nunique()}')
train_peptides
# UniProt 相关蛋白质的UniProt代码,每种蛋白质中通常有几种肽
# Peptide 肽中包含的氨基酸序列
# PeptideAbundance 样品中肽的频率

1.3.从肽水平数据汇总的蛋白表达频率数据 来自train_proteins.csv
print(f'患者人数:{train_protiens["patient_id"].nunique()}')
print(f'月份数:{train_protiens["visit_month"].nunique()}')
print(f'蛋白质数目:{train_protiens["UniProt"].nunique()}')
train_protiens
# NPX 蛋白质在样品中出现的频率
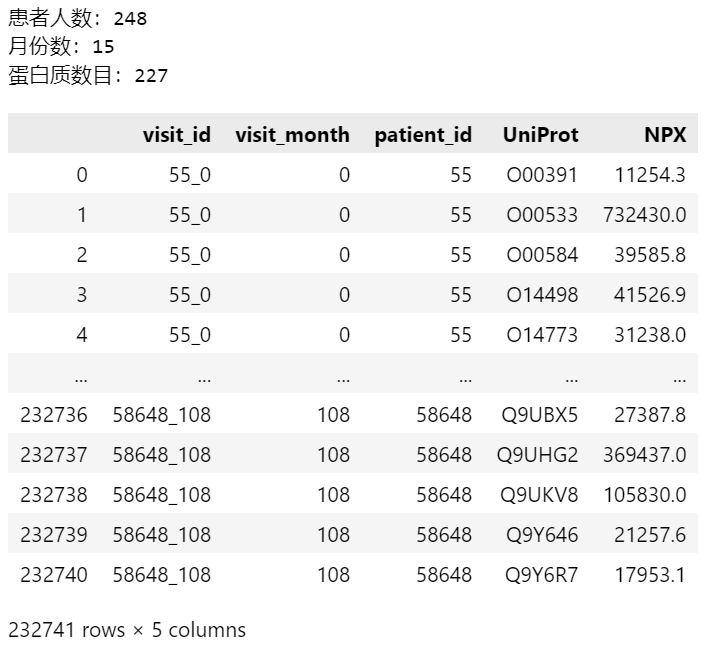
1.4.没有任何相关脑脊液样本的临床记录 来自supplemental_clinical_data
print(f'患者人数:{supplemental_clinical_data["patient_id"].nunique()}')
print(f'月份数:{supplemental_clinical_data["visit_month"].nunique()}')
# 该数据旨在提供有关帕金森病典型进展的其他背景信息,使用与train_clinical_data.csv相同的列。
supplemental_clinical_data
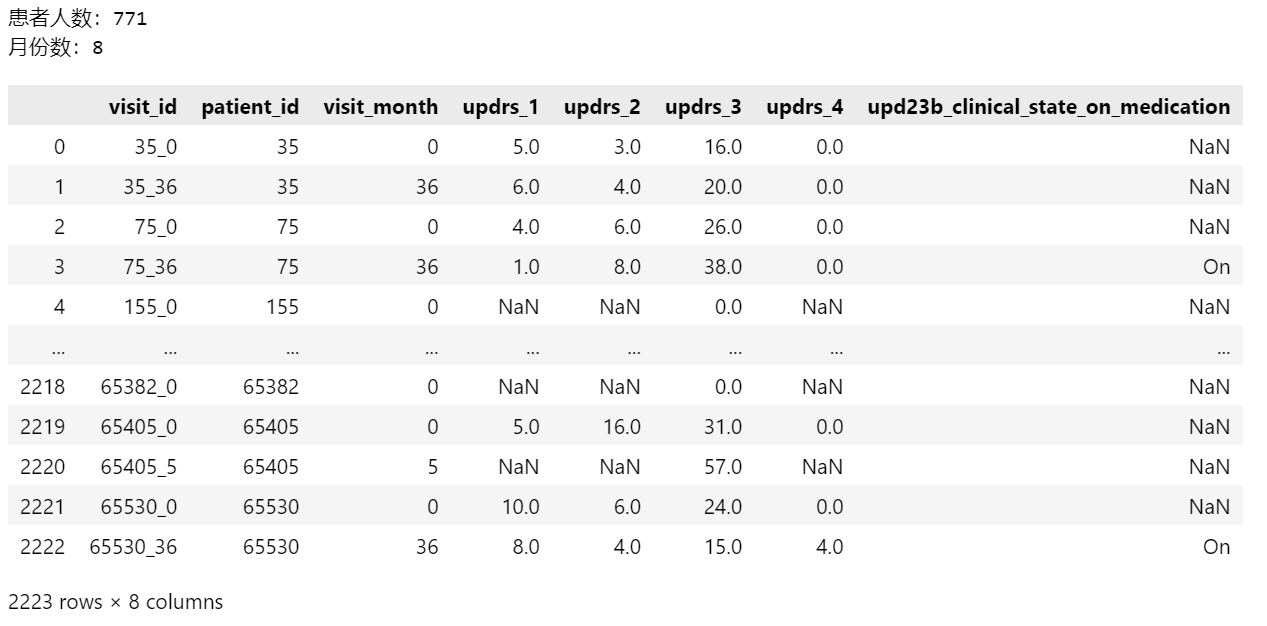
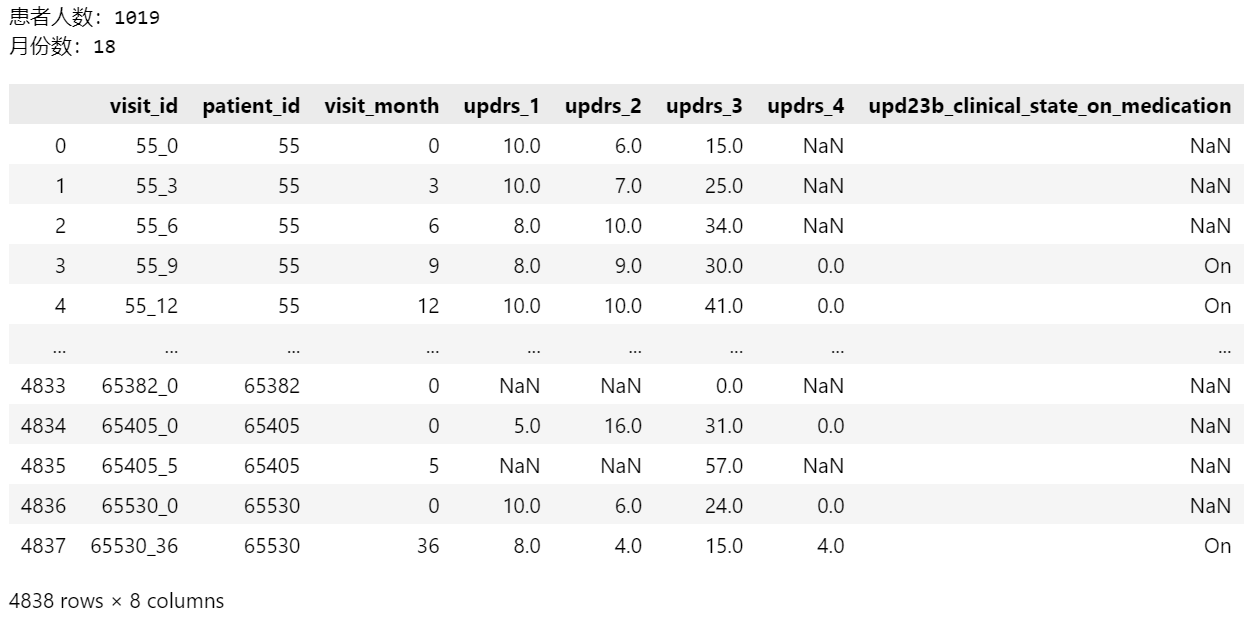
将含脑脊液的的样本与支持样本拼接
combined = pd.concat([train_clinical_data, supplemental_clinical_data]).reset_index(drop=True)
print(f'患者人数:{combined["patient_id"].nunique()}')
print(f'月份数:{combined["visit_month"].nunique()}')
combined
总结
1.train_clinical_data
此数据集表示患者在特定月份的统一帕金森病评定量表1-4部分的得分,其中upd23b_clinical_state_on_medication是类别特征,visit_month和updrs_[1-4]是连续特征。
数据集包含2615行数据,248个患者和17个月份。
2.train_peptides
此数据集表示特定患者在特定月份的脑脊液中所见的肽频率,此数据集基于visit_id连接到train_clinical_data的数据集,其中UniProt与Peptide是类别特征,PeptideAbundance是连续特征。
数据集包含981834行数据,248个患者,227种蛋白质,968种肽。
3.train_proteins
此数据集代表特定患者在特定月份在脑脊液中看到的蛋白质表达频率,此数据集基于visit_id连接到train_clinical_data数据集,其中UniProt是类别特征,NPX是连续特征。
数据集包含232741行数据,248个患者,227种蛋白质。
4.supplemental_clinical_data是补充性临床数据,该数据集代表了补充信息,没有任何来自脑脊液的蛋白质或肽的数据。其中upd23b_clinical_state_on_medication是类别特征,visit_month和updrs_[1-4]是连续特征。
此数据集包含2223行数据,771个患者。
总计
4838次独立访问
1019名患者
18个月份
2.NULL值
from matplotlib import pyplot as plt
import seaborn as sns
# 记录train_clinical_data的缺失信息
train_clinical_data['null_count'] = train_clinical_data.isnull().sum(axis=1)
# 按照null_count字段进行分组,然后计算每组visit_id字段的数量
counts_train_clinical_data = train_clinical_data.groupby('null_count')['visit_id'].count().to_dict()
null_train_clinical_data = {"{} Null Value(s)".format(k) : v for k, v in counts_train_clinical_data.items()}
print(f'train_clinical_data: {null_train_clinical_data}')
# supplemental_clinical_data
supplemental_clinical_data["null_count"] = supplemental_clinical_data.isnull().sum(axis=1)
counts_supplemental_clinical_data = supplemental_clinical_data.groupby("null_count")["visit_id"].count().to_dict()
null_supplemental_clinical_data = {"{} Null Value(s)".format(k) : v for k, v in counts_supplemental_clinical_data.items()}
print(f'supplemental_clinical_data: {null_supplemental_clinical_data}')
# train_protiens
train_protiens["null_count"] = train_protiens.isnull().sum(axis=1)
counts_train_protiens = train_protiens.groupby("null_count")["visit_id"].count().to_dict()
null_train_protiens = {"{} Null Value(s)".format(k) : v for k, v in counts_train_protiens.items()}
print(f'train_protiens: {null_train_protiens}')
# train_peptides
train_peptides["null_count"] = train_peptides.isnull().sum(axis=1)
counts_train_peptides = train_peptides.groupby("null_count")["visit_id"].count().to_dict()
null_train_peptides = {"{} Null Value(s)".format(k) : v for k, v in counts_train_peptides.items()}
print(f'train_peptides: {null_train_peptides}')
fig, axs = plt.subplots(nrows=2, ncols=2, figsize=(15, 15)) # fig 是整个图形的对象,axs 是一个包含了每个子图对象的数组
axs = axs.flatten() # axs = axs.flatten() 是将 axs 数组展平,使得我们可以方便地使用单个索引来访问每个子图
_ = axs[0].pie(
x=list(null_train_clinical_data.values()),
autopct=lambda x: f'{x * sum(null_train_clinical_data.values())/100:.0f} = {x:.2f}',
explode=[0.05] * len(null_train_clinical_data.keys()), # explode 参数表示距离饼图中心的偏移量
labels=null_train_clinical_data.keys(),
colors=sns.color_palette("Set2")[0:len(null_train_clinical_data.keys())],
)
_ = axs[0].set_title("Null Values Per Row in Clinical Data", fontsize=15)
_ = axs[1].pie(
x=list(null_supplemental_clinical_data.values()),
autopct=lambda x: "{:,.0f} = {:.2f}%".format(x * sum(null_supplemental_clinical_data.values())/100, x),
explode=[0.05] * len(null_supplemental_clinical_data.keys()),
labels=null_supplemental_clinical_data.keys(),
colors=sns.color_palette("Set2")[0:len(null_supplemental_clinical_data.keys())],
)
_ = axs[1].set_title("Null Values Per Row in Supplemental Clinical Data", fontsize=15)
_ = axs[2].pie(
x=list(null_train_protiens.values()),
autopct=lambda x: "{:,.0f} = {:.2f}%".format(x * sum(null_train_protiens.values())/100, x),
explode=[0.05] * len(null_train_protiens.keys()),
labels=null_train_protiens.keys(),
colors=sns.color_palette("Set2")[0:len(null_train_protiens.keys())],
)
_ = axs[2].set_title("Null Values Per Row in Protein Data", fontsize=15)
_ = axs[3].pie(
x=list(null_train_peptides.values()),
autopct=lambda x: "{:,.0f} = {:.2f}%".format(x * sum(null_train_peptides.values())/100, x),
explode=[0.05] * len(null_train_peptides.keys()),
labels=null_train_peptides.keys(),
colors=sns.color_palette("Set2")[0:len(null_train_peptides.keys())],
)
_ = axs[3].set_title("Null Values Per Row in Peptide Data", fontsize=15)
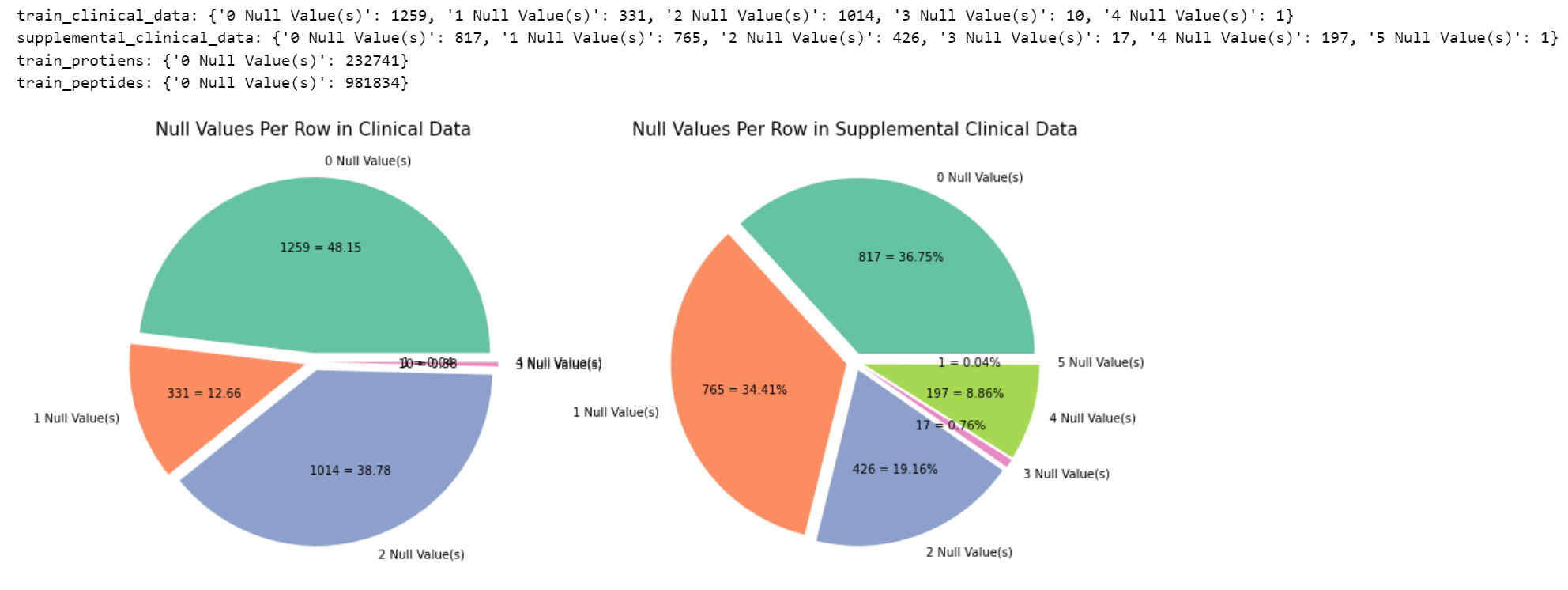
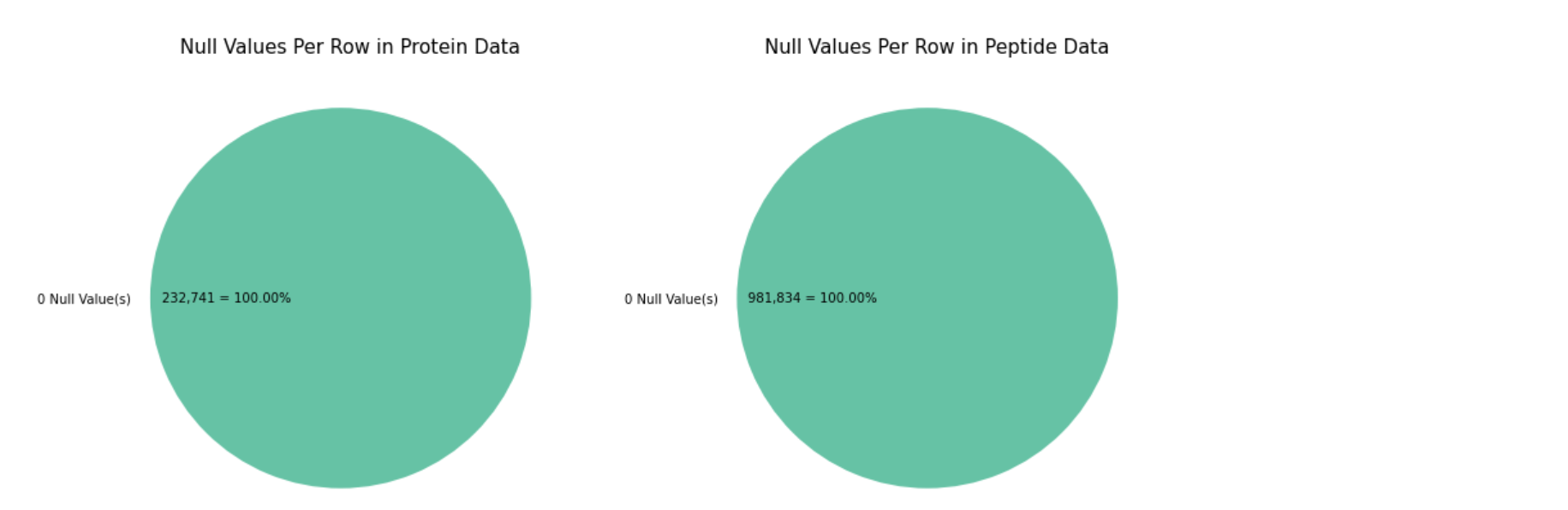
由扇形图可以看到涉及肽和蛋白质的数据无空值,但涉及到临床数据时是有空值的。
接下来看一下train_clinical_data的缺失信息
# 选取所有具有相同非空值数量的行,并将每行中的所有空值求和
# isnull().sum() 函数计算选定行中的所有空值数, .index[:-1] 则返回一个包含除目标列外的所有列索引的列表
null_count_labels = [train_clinical_data[(train_clinical_data["null_count"] == x)].isnull().sum().index[:-1] for x in range(1, 6)]
null_count_values = [train_clinical_data[(train_clinical_data["null_count"] == x)].isnull().sum().values[:-1] for x in range(1, 6)]
fig, axs = plt.subplots(nrows=1, ncols=4, figsize=(20, 5))
fig.suptitle("Null Values for Clinical Data", fontsize=20)
axs = axs.flatten()
for x in range(0, 4):
ax = axs[x]
labels = null_count_labels[x]
_ = sns.barplot(x=labels, y=null_count_values[x], ax=ax)
_ = ax.set_title("Number of Rows With {} Null(s)".format(x + 1), fontsize=15)
_ = ax.set_ylabel("# of Nulls" if x == 0 else "")
_ = ax.set_xlabel("")
_ = ax.set_xticks([z for z in range(len(labels))], labels, rotation=90)
for p in ax.patches:
height = p.get_height()
ax.text(x=p.get_x()+(p.get_width()/2), y=height, s="{:d}".format(int(height)), ha="center")
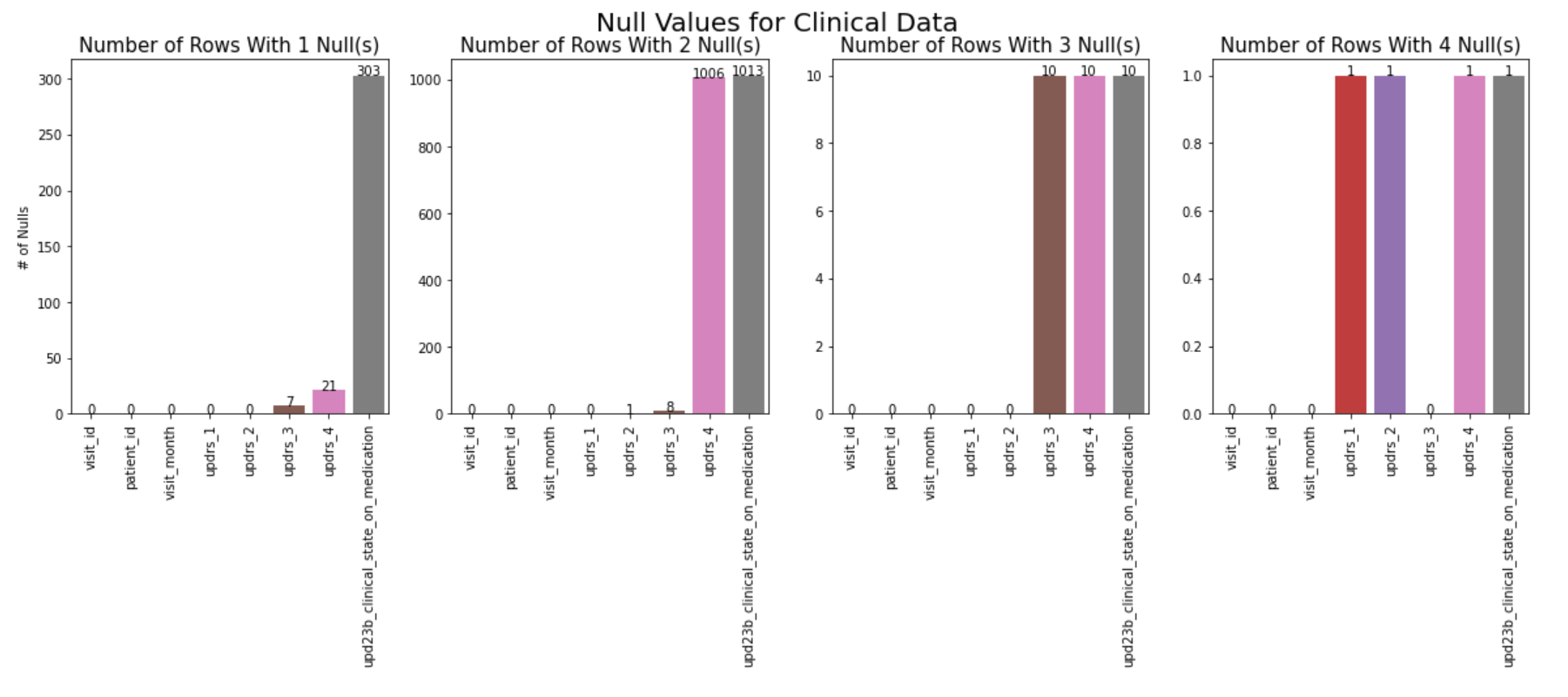
分析
当一行有1个空值时,通常对应于特征upd23b_clinical_state_on_medication,由On或Off组成,该字段的空值无法确定它们是否表示患者停止服药,或者评估是否未能捕捉到患者的服药状态。updrs_3出现了7次,updrs_4出现了21次,根据 Goetz 等人 (2008) 的说法,UPDRS 评估的第 3 部分涉及运动评估,最低得分为 0。UPDRS 评估的第 4 部分涉及运动并发症,同样最低得分为 0。 这些列表明评估未执行。 这一点很重要,因为 0 分表示患者已接受评估并被认为具有正常反应,所以不能做简单的补0操作。
当一行有2个空值时,通常对应于特征upd23b_clinical_state_on_medication和updrs_4,少部分出现在updrs_2和updrs_3,对于 UPDRS 第 2 部分,评估涉及日常生活的运动体验。
有 10 个实例的行包含 3 个空值。 在每个实例中,该行都缺少来自updrs_3、updrs_4和upd23b_clinical_state_on_medication的信息。 同样,缺失值不能假定为 0。
仅出现具有 4 个空值的行的单个实例。 访问时似乎只执行了 UPDRS 第 3 部分评估。 同样,空值不能解释为 0,因为基于 0 的分数表示正常响应。
supplemental_clinical_data的缺失信息
null_count_labels = [supplemental_clinical_data[(supplemental_clinical_data["null_count"] == x)].isnull().sum().index[:-1] for x in range(1, 6)]
null_count_values = [supplemental_clinical_data[(supplemental_clinical_data["null_count"] == x)].isnull().sum().values[:-1] for x in range(1, 6)]
fig, axs = plt.subplots(nrows=1, ncols=4, figsize=(20, 5))
fig.suptitle("Null Values for Supplemental Data", fontsize=20)
axs = axs.flatten()
for x in range(0, 4):
ax = axs[x]
labels = null_count_labels[x]
_ = sns.barplot(x=labels, y=null_count_values[x], ax=ax)
_ = ax.set_title("Number of Rows With {} Null(s)".format(x + 1), fontsize=15)
_ = ax.set_ylabel("# of Nulls" if x == 0 else "")
_ = ax.set_xlabel("Feature")
_ = ax.set_xticks([z for z in range(len(labels))], labels, rotation=90)
for p in ax.patches:
height = p.get_height()
ax.text(x=p.get_x()+(p.get_width()/2), y=height, s="{:d}".format(int(height)), ha="center")
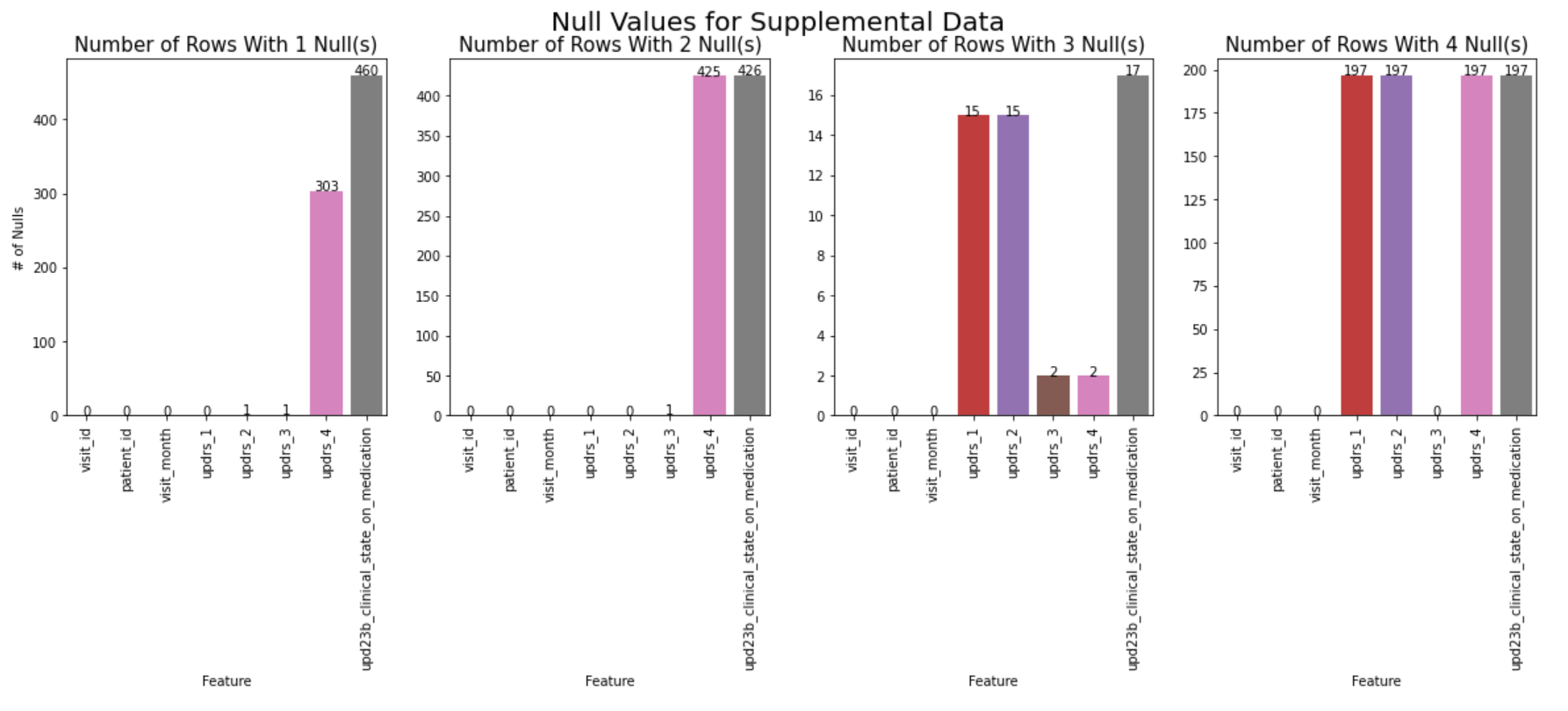
分析
当一行中有1个空值或2个空值时,它会出现在 updrs_4 和 upd23b_clinical_state_on_medication 中。
当存在3个空值时,趋势与train_clinical_data略有不同。 在这种情况下,与train_clinical_data相比,补充数据更有可能缺少updrs_1和updrs_2。 最后,当有4个缺失值时,我们在updrs_1、updrs_2、updrs_4 和upd23b_clinical_state_on_medication 中看到相同的缺失值,补充数据中 4 个空值行的数量明显多于临床数据。
处理空值时必须小心:
是否应该使用空值来表示错过评估,或者是否可以将它们设置为另一个值,这并不明显。
- 对于 UPDRS 评估,将值设置为 0 可能是错误的,因为这表示“正常”结果。
- 对于
upd23b_clinical_state_on_medication特性,唯一有效的设置是On或Off,因此 null 的影响是未定义的。
3.重复行
重复可能会影响我们的学习方法,导致对重复信息的预测偏差。
import numpy as np
titles = ["Peptide Data", "Protein Data", "Clinical Data", "Supplemental Data"]
value_counts = []
# pivot_table 透视表
# 这行代码使用了 pandas library 中的 pivot_table 函数,
# 将 train_peptides dataframe 以 UniProt, Peptide 和 PeptideAbundance 列作为 index,调用 size 函数,
# 得到一个 Series 对象,其中每行表示相应的行组合在 train_peptides dataframe 中出现的次数
duplicates = train_peptides.pivot_table(index=[
'UniProt', 'Peptide', 'PeptideAbundance',
], aggfunc="size")
unique, counts = np.unique(duplicates, return_counts=True)
value_counts.append(dict(zip(unique, counts)))
duplicates = train_protiens.pivot_table(index=[
'UniProt', 'NPX',
], aggfunc="size")
unique, counts = np.unique(duplicates, return_counts=True)
value_counts.append(dict(zip(unique, counts)))
duplicates = train_clinical_data.pivot_table(index=[
'visit_month', 'updrs_1', 'updrs_2', 'updrs_3', 'updrs_4', 'upd23b_clinical_state_on_medication'
], aggfunc="size")
unique, counts = np.unique(duplicates, return_counts=True)
value_counts.append(dict(zip(unique, counts)))
duplicates = supplemental_clinical_data.pivot_table(index=[
'visit_month', 'updrs_1', 'updrs_2', 'updrs_3', 'updrs_4', 'upd23b_clinical_state_on_medication'
], aggfunc="size")
unique, counts = np.unique(duplicates, return_counts=True)
value_counts.append(dict(zip(unique, counts)))
fig, axs = plt.subplots(nrows=1, ncols=4, figsize=(20, 4))
axs = axs.flatten()
for x in range(4):
ax = axs[x]
_ = sns.barplot(x=list(value_counts[x].keys())[1:], y=list(value_counts[x].values())[1:], ax=ax)
_ = ax.set_title("Duplicate Counts in {}".format(titles[x], fontsize=15))
_ = ax.set_ylabel("Count")
_ = ax.set_xlabel("Number of Times Row is Duplicated")
for p in ax.patches:
height = p.get_height()
ax.text(x=p.get_x()+(p.get_width()/2), y=height, s="{:d}".format(int(height)), ha="center")
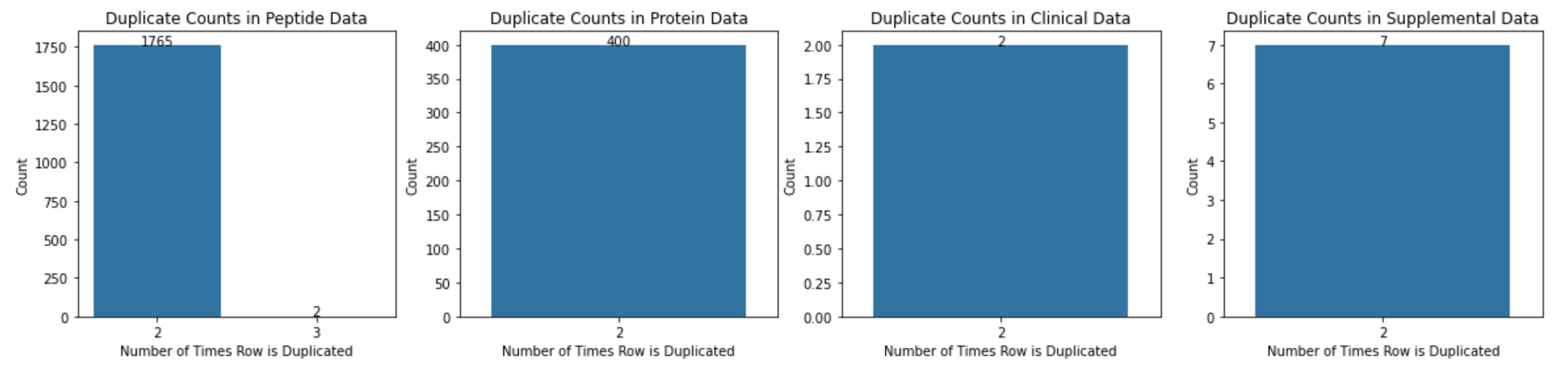
关于重复行的主要观察
- 就原始副本而言:
- 在临床数据中:
- 同一行数据出现两次的情况有 2 次。
- 重复数据占所有临床数据的 0.15%。
- 在补充数据中:
- 同一行数据出现两次的情况有 7 次。
- 重复数据占所有补充数据的 0.63%。
- 在蛋白质数据中:
- 同一行数据出现两次的情况有 400 个。
- 重复项占所有蛋白质数据的 0.35%。
- 在肽数据中:
- 同一行数据出现两次的情况有 1,765 个。
- 同一行数据出现 3 次的情况有 2 次。
- 重复项占所有肽数据的 0.36%。
- 在临床数据中:
- 总体而言,对于临床和补充数据,重复数据可能影响很小或没有影响。
4.统计分析
首先,让我们将临床数据与补充数据进行比较,看看我们有什么样的差异。
print('train_clinical_data')
features = ['visit_month', 'updrs_1', 'updrs_2', 'updrs_3', 'updrs_4',]
# .T: 转置整个数据表,行列互换。
# .style: 对 DataFrame 进行美化,返回一个 Styler 对象,进而可以链式描述所需的格式。
# 对 mean 的 cells 标成条形图
# 对 std 的 cells 进行 cmap='Reds' 颜色的背景渐变,即数字越大的单元格颜色越深
# 对 50 percentile 的 cells 进行 cmap='coolwarm' 颜色的背景渐变,即数字越大/小的单元格颜色越深/浅
train_clinical_data[features].describe().T.style.bar(subset=['mean'], color='#7BCC70')\
.background_gradient(subset=['std'], cmap='Reds')\
.background_gradient(subset=['50%'], cmap='coolwarm')
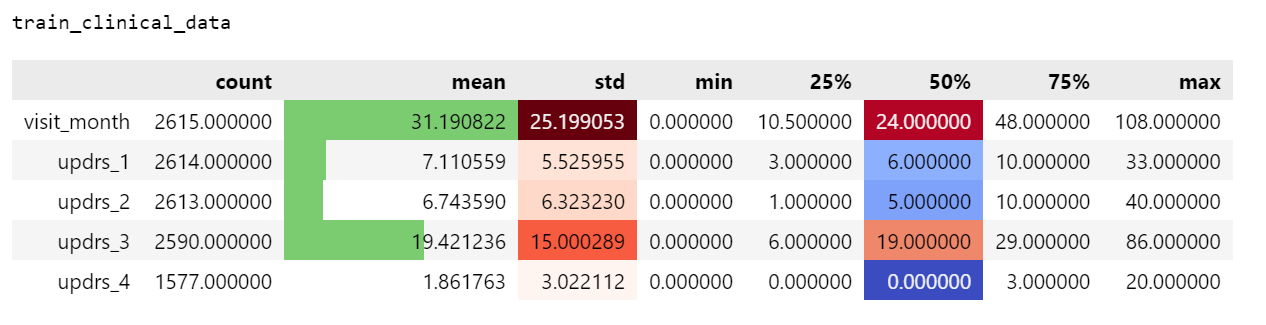
print('supplemental_clinical_data')
features = ['visit_month', 'updrs_1', 'updrs_2', 'updrs_3', 'updrs_4',]
supplemental_clinical_data[features].describe().T.style.bar(subset=['mean'], color='#7BCC70')\
.background_gradient(subset=['std'], cmap='Reds')\
.background_gradient(subset=['50%'], cmap='coolwarm')
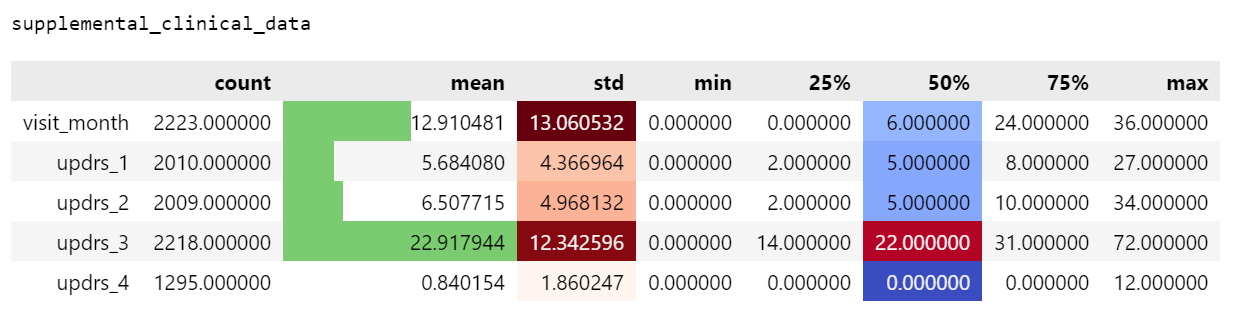
补充数据显示就诊主要发生在 0 到 36 个月之间,而临床数据显示就诊发生在 0 到 108 个月之间。
接下来比较一下按月就诊的分布情况:
train_clinical_data["origin"] = "Clincial Data"
supplemental_clinical_data["origin"] = "Supplemental Data"
combined = pd.concat([train_clinical_data, supplemental_clinical_data]).reset_index(drop=True)
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(15, 5))
sns.set_style('darkgrid')
_ = sns.histplot(data=combined, x="visit_month", hue="origin", kde=True, ax=ax, element="step")
_ = ax.set_title("Visit Month by Data Source", fontsize=15)
_ = ax.set_ylabel("Count")
_ = ax.set_xlabel("Visit Month")
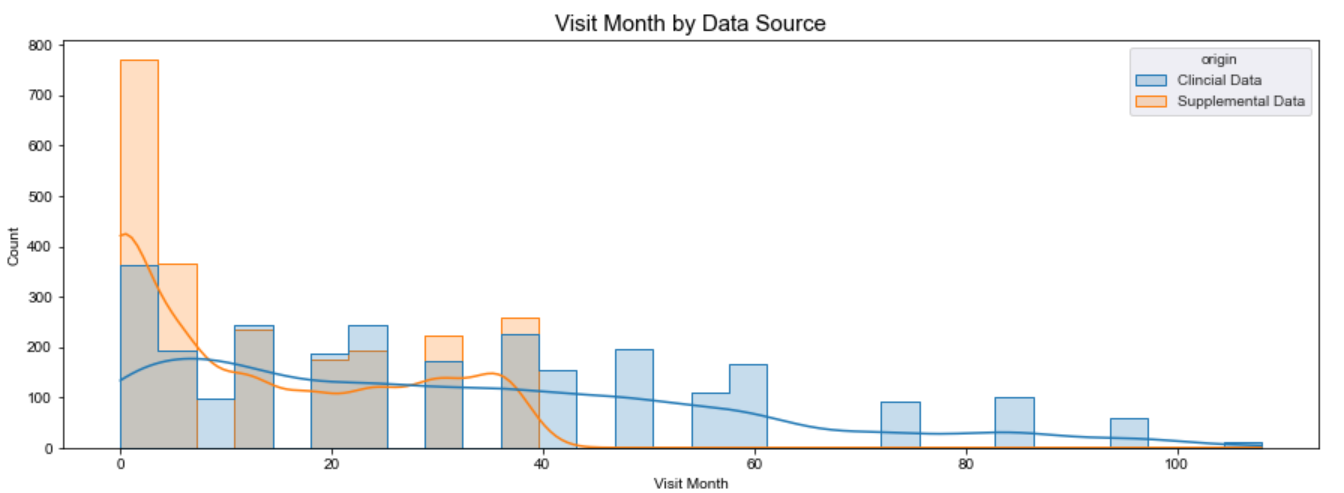
我们还可以进行快速目视检查,看看临床数据和补充数据在 UPDRS 评分方面是否存在差异。 对于下图,趋势线是核密度估计值,因此趋势线考虑了原始计数的差异。
train_clinical_data["origin"] = "Clincial Data"
supplemental_clinical_data["origin"] = "Supplemental Data"
combined = pd.concat([train_clinical_data, supplemental_clinical_data]).reset_index(drop=True)
features = ["updrs_1", "updrs_2", "updrs_3", "updrs_4"]
labels = ["UPDRS Part 1", "UPDRS Part 2", "UPDRS Part 3", "UPDRS Part 4"]
fig, axs = plt.subplots(nrows=4, ncols=1, figsize=(15, 25))
sns.set_style('darkgrid')
axs = axs.flatten()
sns.set_style('darkgrid')
for x, feature in enumerate(features):
ax = axs[x]
_ = sns.histplot(data=combined, x=feature, hue="origin", kde=True, ax=ax, element="step")
_ = ax.set_title("{} Scores by Data Source".format(labels[x]), fontsize=15)
_ = ax.set_ylabel("Count")
_ = ax.set_xlabel("{} Score".format(labels[x]))
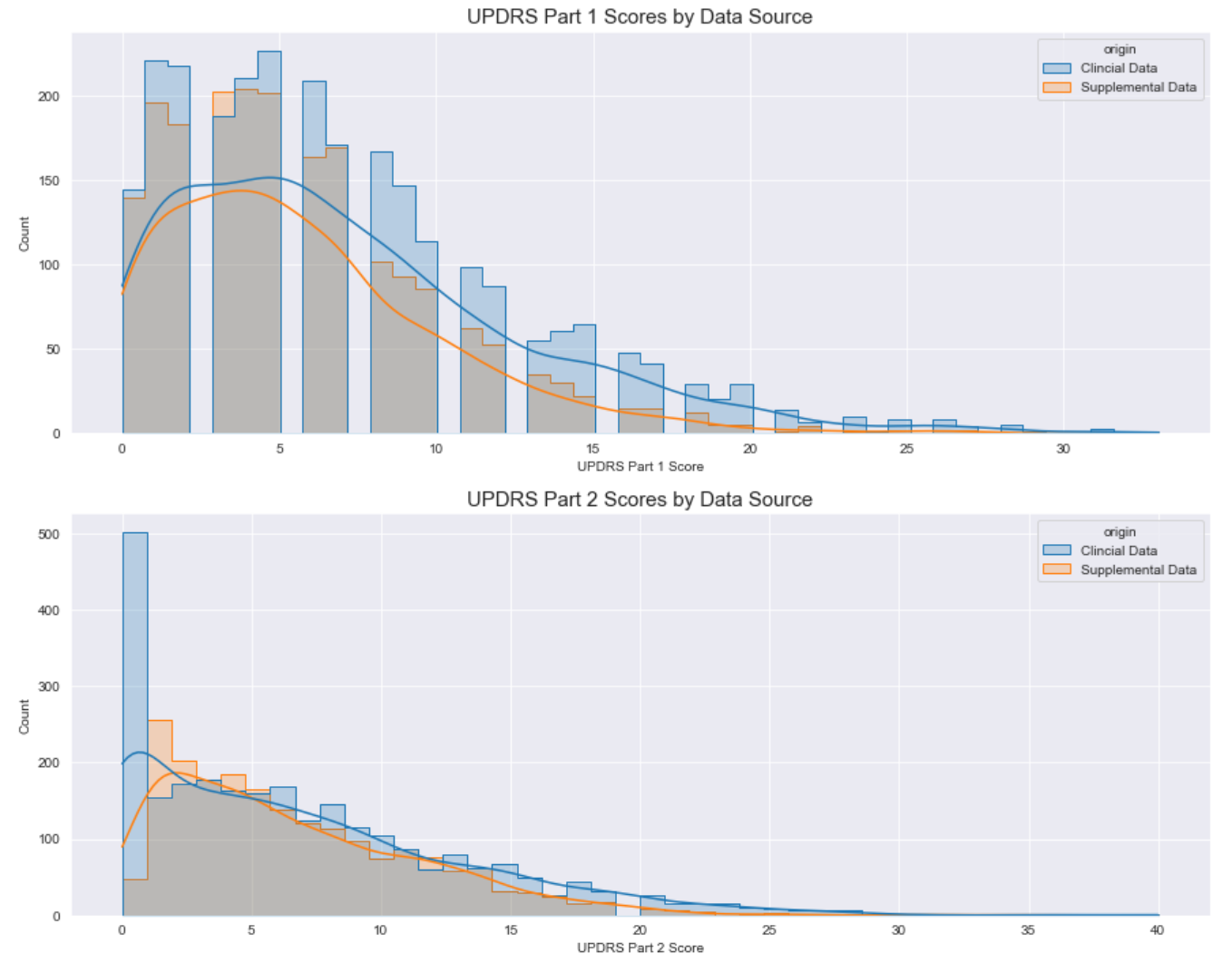
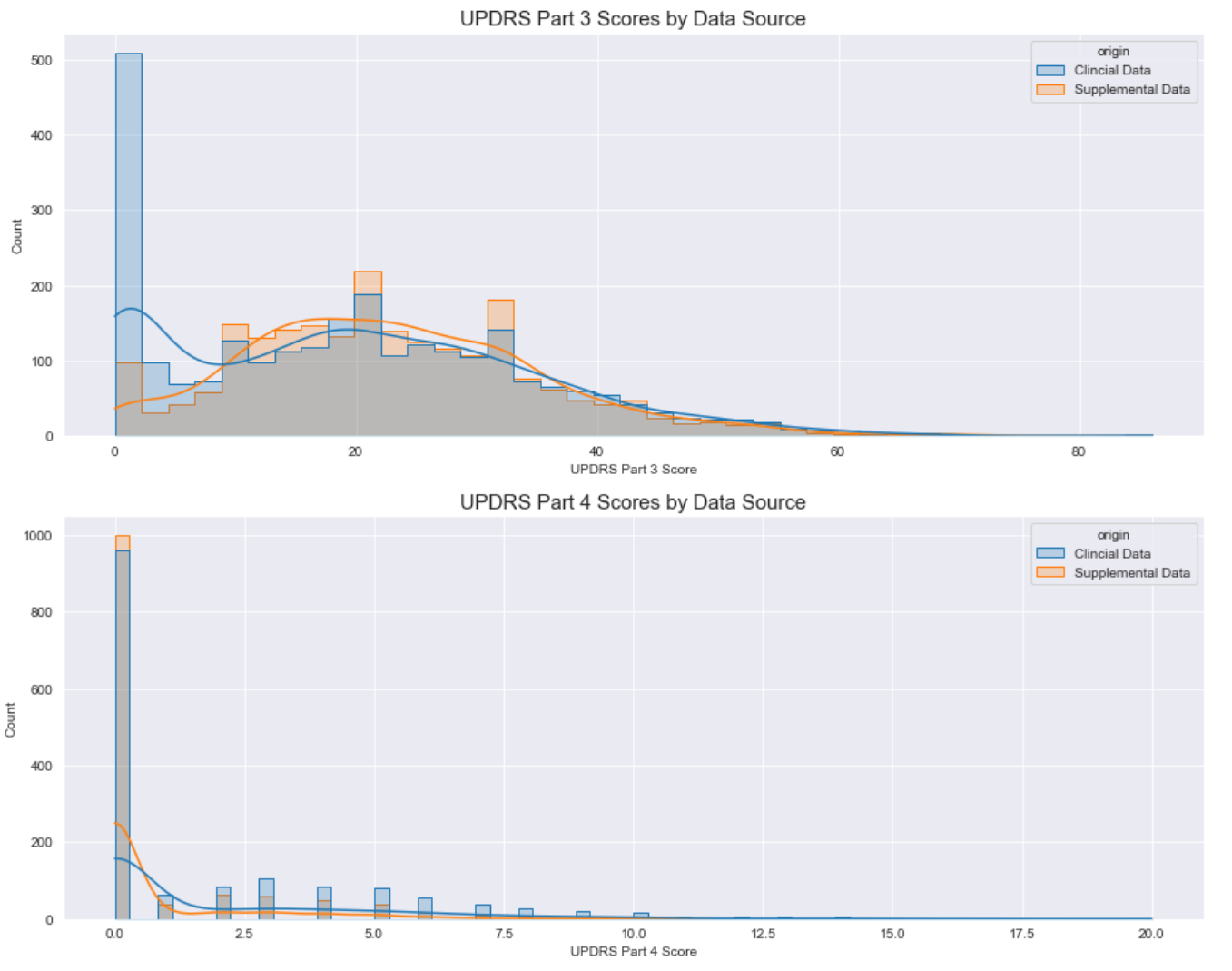
UPDRS 第 1 部分和第 4 部分分数在临床和补充数据源之间的分布似乎非常相似。
我们可以通过执行对抗性验证来粗略衡量临床数据和补充数据之间的差异。 对抗性验证的目标是查看分类器是否可以区分两个数据集。 我们将使用 ROC AUC 分数来告知我们差异。 如果两组看起来非常相似,分类器将无法区分它们,因此 ROC AUC 分数将为 0.5。 如果它们很容易区分, 那么 ROC AUC 分数将接近 1。
from sklearn.model_selection import KFold
from sklearn.metrics import roc_auc_score
from lightgbm import LGBMClassifier
from lightgbm import early_stopping, log_evaluation
from sklearn.metrics import confusion_matrix
from sklearn.metrics import roc_curve
from sklearn.preprocessing import LabelEncoder
# 打标签
train_clinical_data['origin'] = 0
supplemental_clinical_data['origin'] = 1
combined = pd.concat([train_clinical_data, supplemental_clinical_data]).reset_index(drop=True)
features = ['visit_month', 'updrs_1', 'updrs_2', 'updrs_3', 'updrs_4', 'upd23b_clinical_state_on_medication']
le = LabelEncoder() # LabelEncoder 将分类变量映射为从 0 开始连续的整数,一个类别对应一个标签
combined['upd23b_clinical_state_on_medication'] = le.fit_transform(combined['upd23b_clinical_state_on_medication'])
n_folds = 5
skf = KFold(n_splits=n_folds, random_state=2023, shuffle=True)
train_oof_preds = np.zeros((combined.shape[0]))
train_oof_probas = np.zeros((combined.shape[0]))
for fold, (train_index, test_index) in enumerate(skf.split(combined, combined['origin'])):
# 对 k-fold 交叉验证器生成的每一组训练和测试集的索引进行循环
print(f'-------> Fold {fold + 1} <--------')
x_train, x_valid = pd.DataFrame(combined.iloc[train_index]), pd.DataFrame(combined.iloc[test_index])
y_train, y_valid = combined["origin"].iloc[train_index], combined["origin"].iloc[test_index]
# 在整个数据集中取出了需要使用的特征进行后续的训练和测试
x_train_features = pd.DataFrame(x_train[features])
x_valid_features = pd.DataFrame(x_valid[features])
model = LGBMClassifier(
random_state=2023, # 模型中用于产生随机数的种子
objective="binary", # 模型采用二分类任务,优化目标为最大化 AUC
metric="auc", # 模型在训练过程中衡量效果的指标是 AUC
n_jobs=-1, # 并行运算线程数,-1 表示使用所有 CPU 核心
n_estimators=2000, # 分类器中选择的树形成员数目
verbose=-1, # 控制输出消息的详细程度。-1 表示不输出
max_depth=3, # 单棵树的最大深度
)
model.fit(
x_train_features[features], # 训练集的特征数据
y_train, # 训练集的目标变量
eval_set=[(x_valid_features[features], y_valid)], # 测试集和标签
callbacks=[ # 在模型 fit 的过程中执行的操作
early_stopping(50, verbose=False), # 函数基于评价指标的变化来判断模型是否过拟合,并在 50 棵树后没有进一步提升时停止训练
log_evaluation(2000), # 在训练过程中每隔 2000 棵树记录下模型在验证集上的评价指标
]
)
oof_preds = model.predict(x_valid_features[features]) # 方法返回一个类别的预测结果
oof_probas = model.predict_proba(x_valid_features[features])[:,1] # predict_proba 方法返回类别为正类的概率值,而不是简单的类别
train_oof_preds[test_index] = oof_preds
train_oof_probas[test_index] = oof_probas
print(": AUC ROC = {}".format(roc_auc_score(y_valid, oof_probas)))
auc_vanilla = roc_auc_score(combined["origin"], train_oof_probas)
fpr, tpr, _ = roc_curve(combined["origin"], train_oof_probas)
print("--> Overall results for out of fold predictions")
print(": AUC ROC = {}".format(auc_vanilla))
confusion = confusion_matrix(combined["origin"], train_oof_preds)
fig, axs = plt.subplots(nrows=1, ncols=2, figsize=(15, 8))
labels = ["Clincial Data", "Supplemental Data"]
_ = sns.heatmap(confusion, annot=True, fmt=",d", ax=axs[0], xticklabels=labels, yticklabels=labels)
_ = axs[0].set_title("Confusion Matrix (@ 0.5 Probability)", fontsize=15)
_ = axs[0].set_ylabel("Actual Class")
_ = axs[0].set_xlabel("Predicted Class")
_ = sns.lineplot(x=[0, 1], y=[0, 1], linestyle="--", label="Indistinguishable Datasets", ax=axs[1])
_ = sns.lineplot(x=fpr, y=tpr, ax=axs[1], label="Adversarial Validation Classifier")
_ = axs[1].set_title("ROC Curve", fontsize=15)
_ = axs[1].set_xlabel("False Positive Rate")
_ = axs[1].set_ylabel("True Positive Rate")
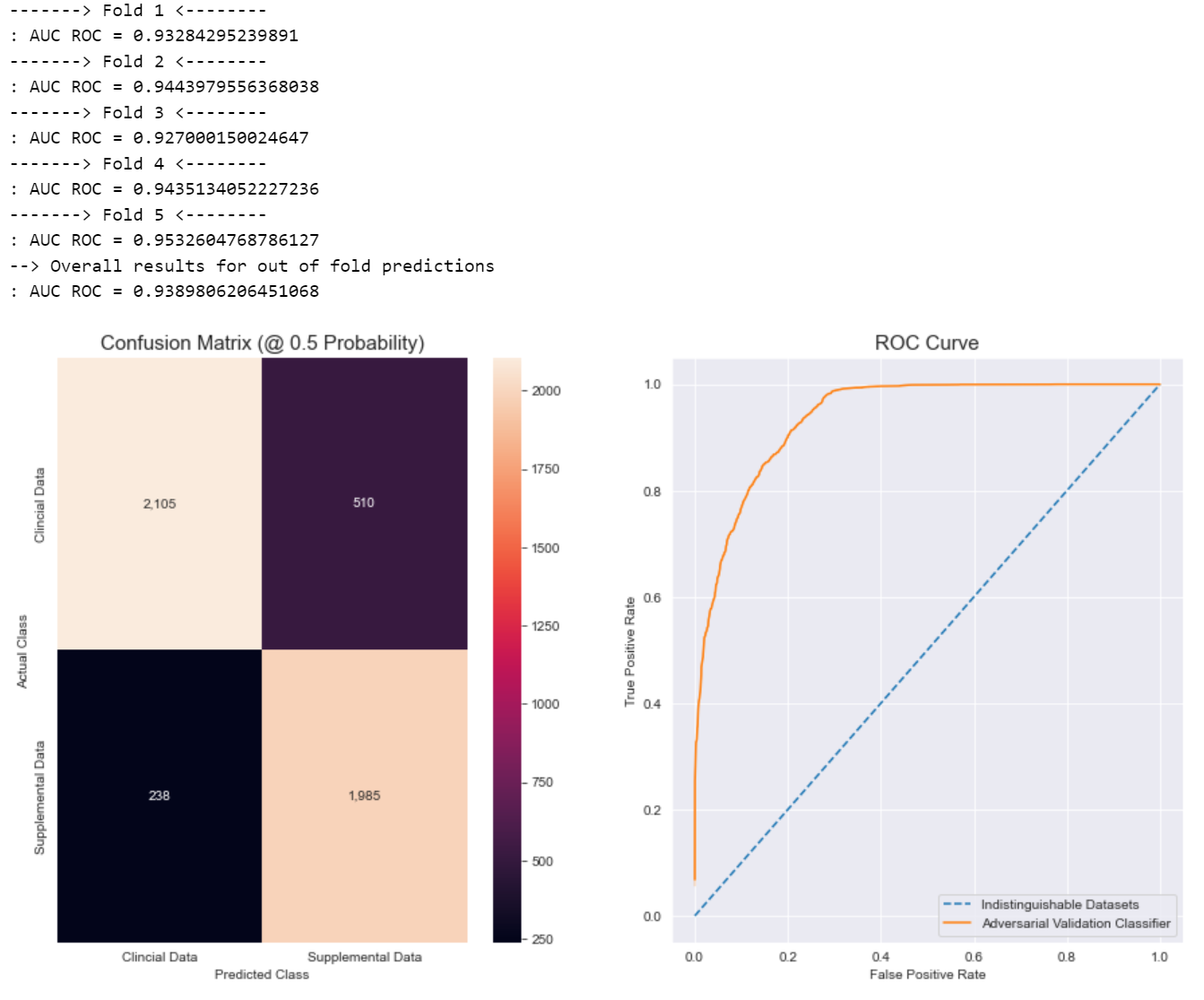
AUC ROC 分数为 0.939,我们可以看到分类器可以轻松区分两个数据集。 这表明它们在本质上非常不同,正如比赛的一部分所指出的那样。 混合这两个数据集时应谨慎,因为它们在本质上非常不同。
蛋白质数据
print('Protein Data')
train_protiens[["NPX"]].describe().T.style.bar(subset=['mean'], color='#7BCC70')\
.background_gradient(subset=['std'], cmap='Reds')\
.background_gradient(subset=['50%'], cmap='coolwarm')

fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(15, 5))
sns.set_style('darkgrid')
# sns.kdeplot() 是一个在 seaborn 库中用于绘制单变量分布的函数。它接收单个一维数组的数据作为输入,并创建表示该数组数据分布的 Kernel Density Estimate(KDE)图
_ = sns.kdeplot(train_protiens["NPX"], shade=True, color="r", ax=ax, label="Normalized Protein Expression", log_scale=True)
_ = ax.set_title("Logarithmic Normalized Protein Expression (Kernel Density Estimate)", fontsize=15)
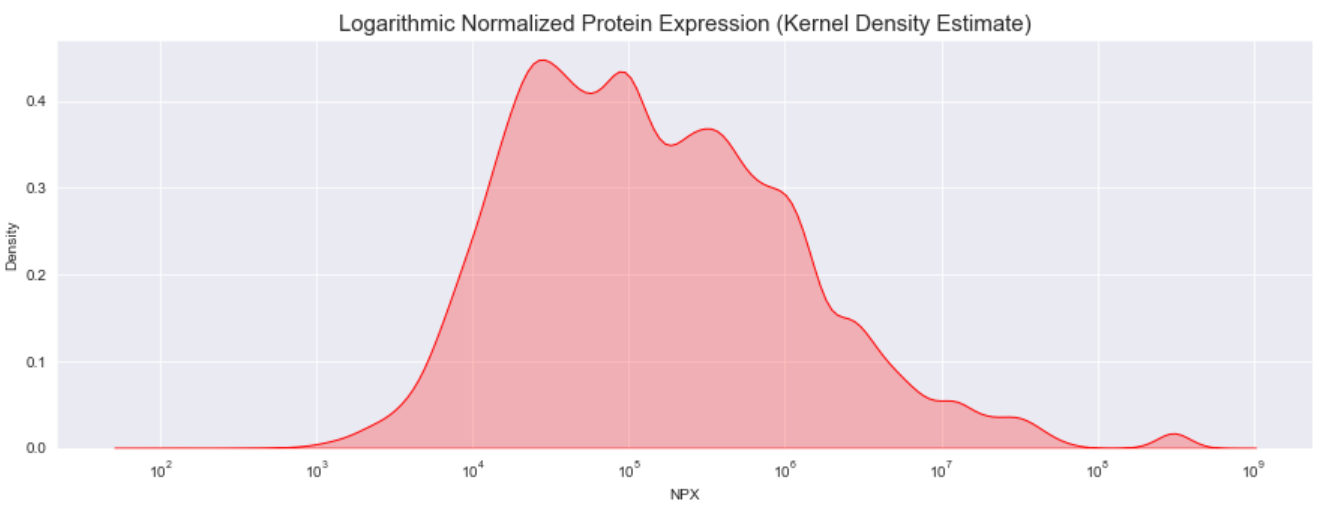
正如我们所见,实际的蛋白质表达频率存在很多差异。 我们将在下面的第 2 节中更多地研究各种蛋白质的分布及其与 UPDRS 分数的关联。 目前,我们的主要观察结果是标准化的蛋白质表达是高度可变的,如特征的最小值、最大值和标准差所示。
肽数据
train_peptides[["PeptideAbundance"]].describe().T.style.bar(subset=['mean'], color='#7BCC70')\
.background_gradient(subset=['std'], cmap='Reds')\
.background_gradient(subset=['50%'], cmap='coolwarm')

同样,我们看到肽的丰度存在很大差异。 最小值、最大值和标准差告诉我们,肽丰度可能会因我们正在查看的特定肽而有很大差异。 同样,我们可以绘制核密度估计值,让我们了解大部分值存在的位置。
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(15, 5))
sns.set_style('darkgrid')
_ = sns.kdeplot(train_peptides["PeptideAbundance"], shade=True, color="r", ax=ax, label="Peptide Abundance", log_scale=True)
_ = ax.set_title("Logarithmic Peptide Abundance (Kernel Density Estimate)", fontsize=15)
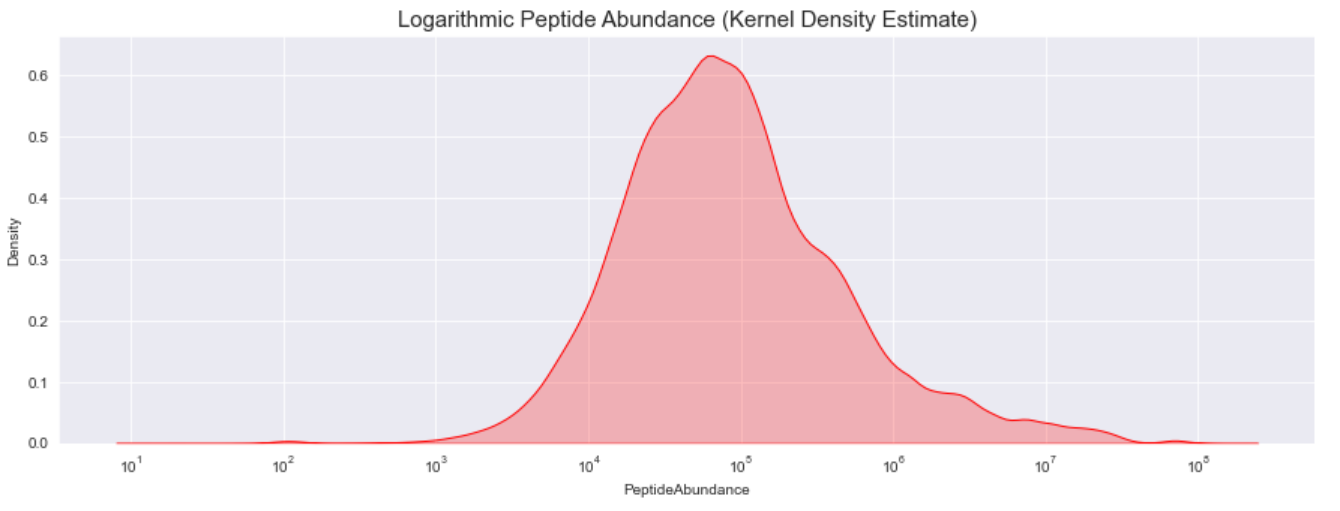
在下面,我们将再次查看肽数据 - 特别是肽序列以及它们与 UPDRS 分数的关系。
关于统计细分的主要观察
- 临床数据和补充数据有非常不同的月份范围:
- 这是通过查看它们的统计属性、直方图和对抗性验证来证实的。
- 蛋白质表达数据具有广泛的价值,因此需要进一步细分为子组以提供信息。
- 肽丰度频率具有广泛的值,因此需要进一步细分为子组以提供信息。
5.特征工程
访问月份会对所有不同的数据集产生影响,并随后通过我们拥有的许多不同特征产生影响。 让我们依次看看它们。
访问月与UPDRS
对于每个访问月,我们都会观察到目标特征 - UPDRS分数。 根据 Holden 等人 (2018) 的说法,UPDRS每个部分的发现都取决于患者是否正在服药。 我们应该将UPDRS评分观察结果细分为服用药物的组和未服用药物的组。 出于此探索的目的,在有关药物状态的临床数据中发现的空值将被视为off。
train_clincial_data_copy = train_clinical_data.copy()
train_clincial_data_copy["upd23b_clinical_state_on_medication"] = train_clincial_data_copy["upd23b_clinical_state_on_medication"].fillna("Off")
fig, axs = plt.subplots(nrows=4, ncols=1, figsize=(15, 25))
sns.set_style('darkgrid')
axs = axs.flatten()
for x, feature in enumerate(["updrs_1", "updrs_2", "updrs_3", "updrs_4"]):
ax = axs[x]
data = train_clincial_data_copy[(train_clincial_data_copy["upd23b_clinical_state_on_medication"] == "Off")]
_ = sns.boxplot(data=data, x="visit_month", y=feature, ax=ax)
_ = sns.pointplot(data=data, x="visit_month", y=feature, color="r", ci=None, linestyles=[":"], ax=ax)
_ = ax.set_title("UPDRS Part {} Scores by Month while OFF Medication".format(x+1), fontsize=15)
_ = ax.set_xlabel("Visit Month")
_ = ax.set_ylabel("Score")
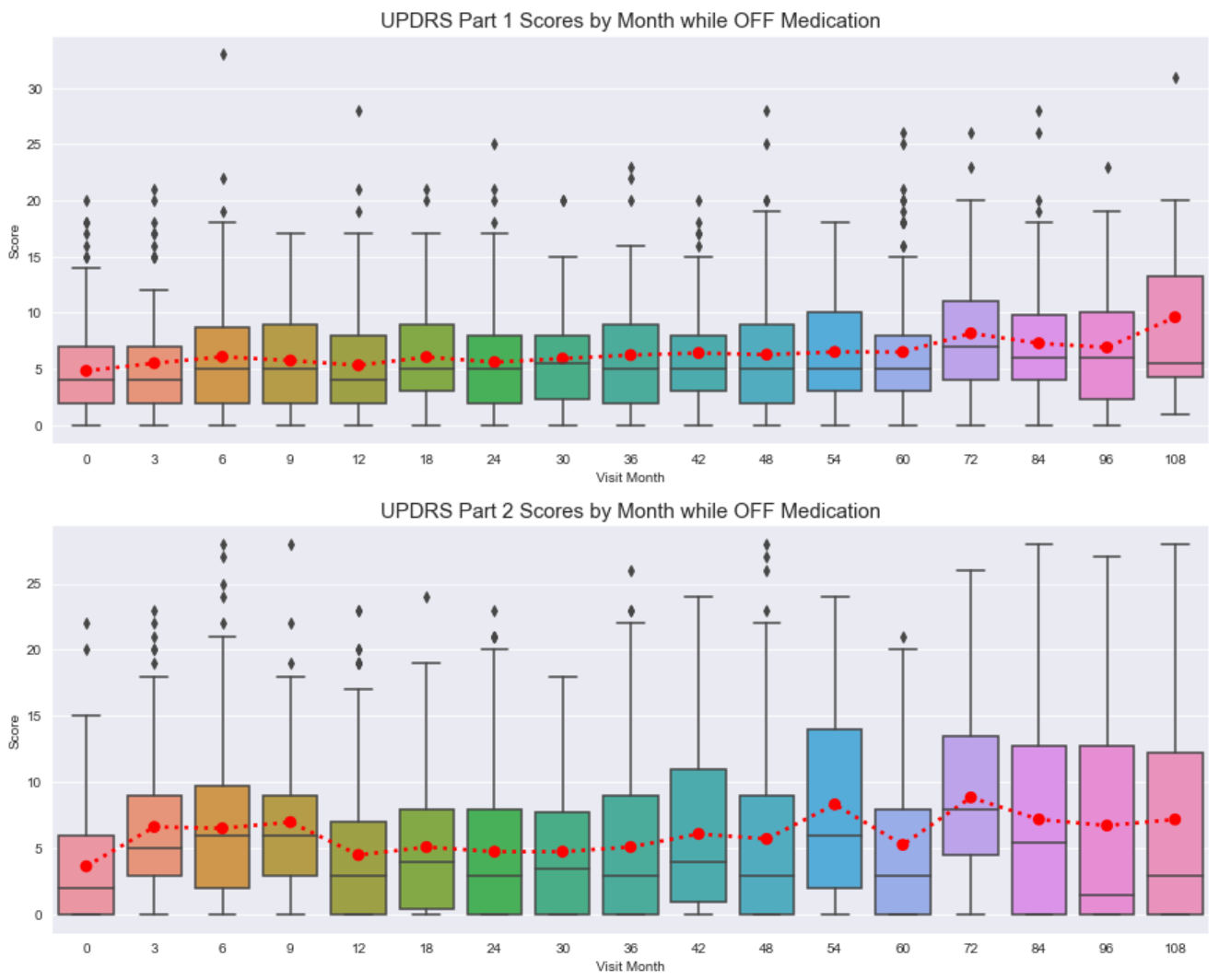
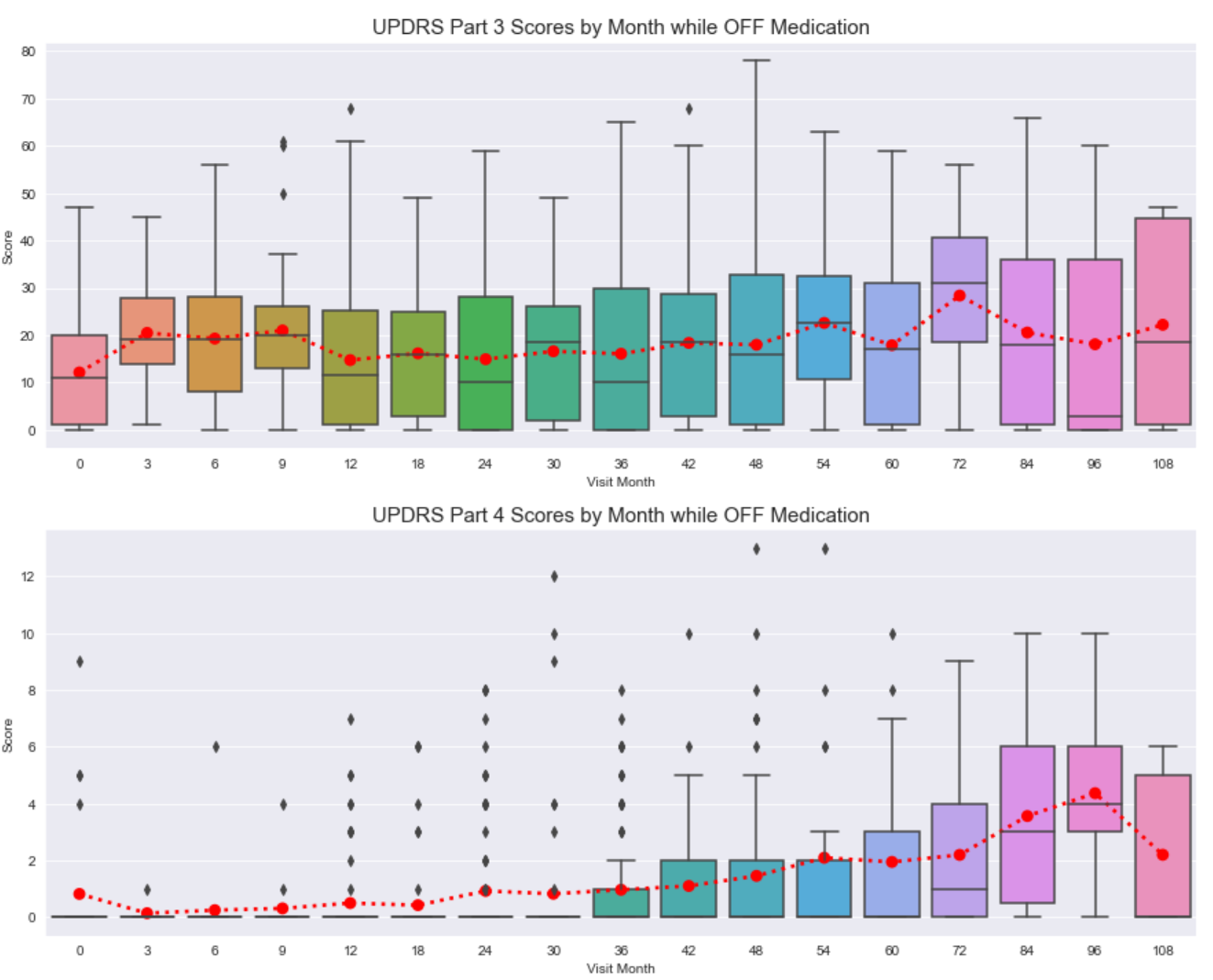
停药时的一些一般观察:
- 每个 UPDRS 部分及其各自的访问月份之间存在大量差异和异常值。
- 总的来说,在 UPDRS 第 1 至第 3 部分中,分数的趋势线保持相对平稳。
- 对于 UPDRS 第 4 部分,我们看到分数逐渐增加。
train_clincial_data_copy = train_clinical_data.copy()
train_clincial_data_copy["upd23b_clinical_state_on_medication"] = train_clincial_data_copy["upd23b_clinical_state_on_medication"].fillna("Off")
fig, axs = plt.subplots(nrows=4, ncols=1, figsize=(15, 25))
sns.set_style('darkgrid')
axs = axs.flatten()
for x, feature in enumerate(["updrs_1", "updrs_2", "updrs_3", "updrs_4"]):
ax = axs[x]
data = train_clincial_data_copy[(train_clincial_data_copy["upd23b_clinical_state_on_medication"] == "On")]
_ = sns.boxplot(data=data, x="visit_month", y=feature, ax=ax)
_ = sns.pointplot(data=data, x="visit_month", y=feature, color="r", ci=None, linestyles=[":"], ax=ax)
_ = ax.set_title("UPDRS Part {} Scores by Month while ON Medication".format(x+1), fontsize=15)
_ = ax.set_xlabel("Visit Month")
_ = ax.set_ylabel("Score")
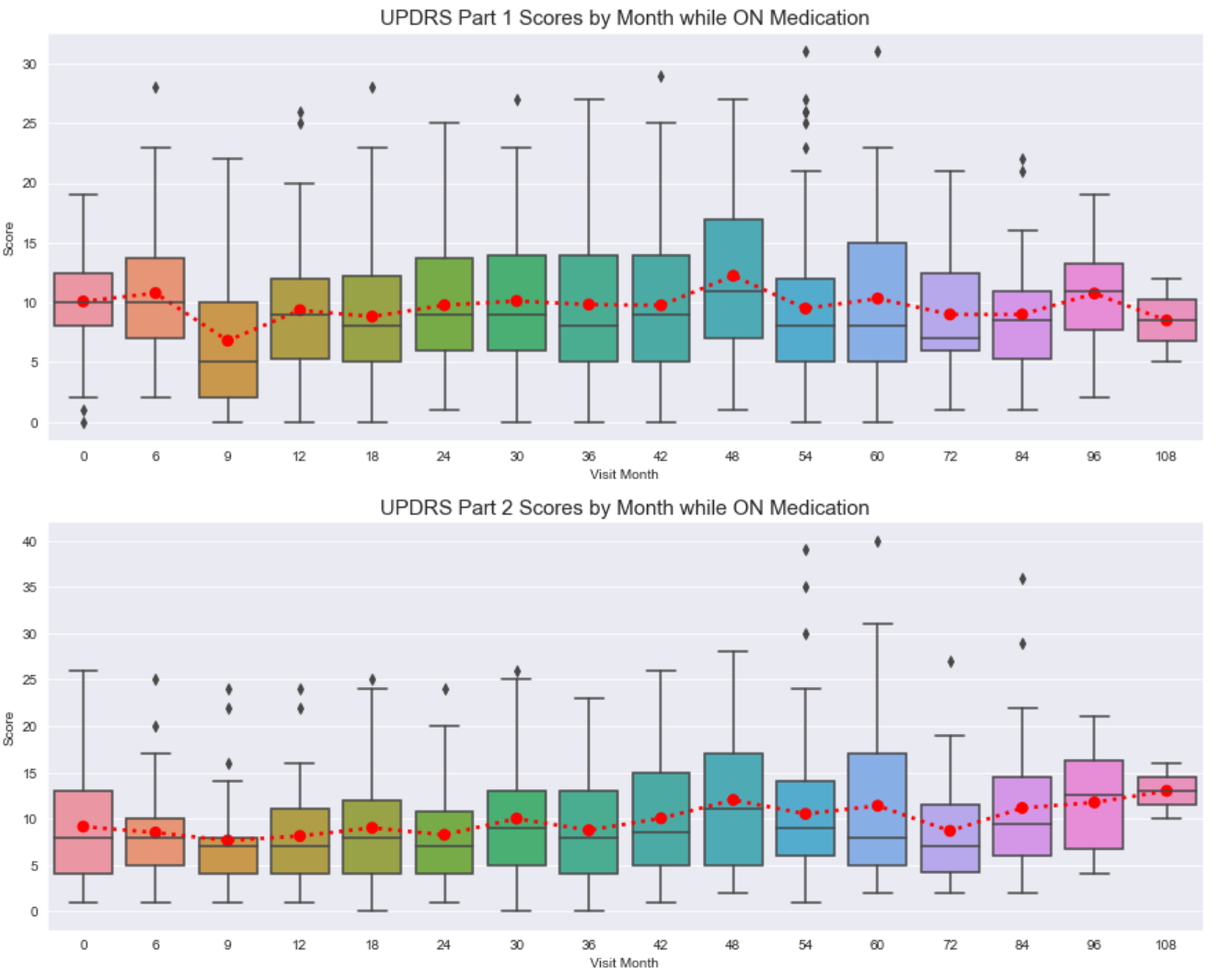
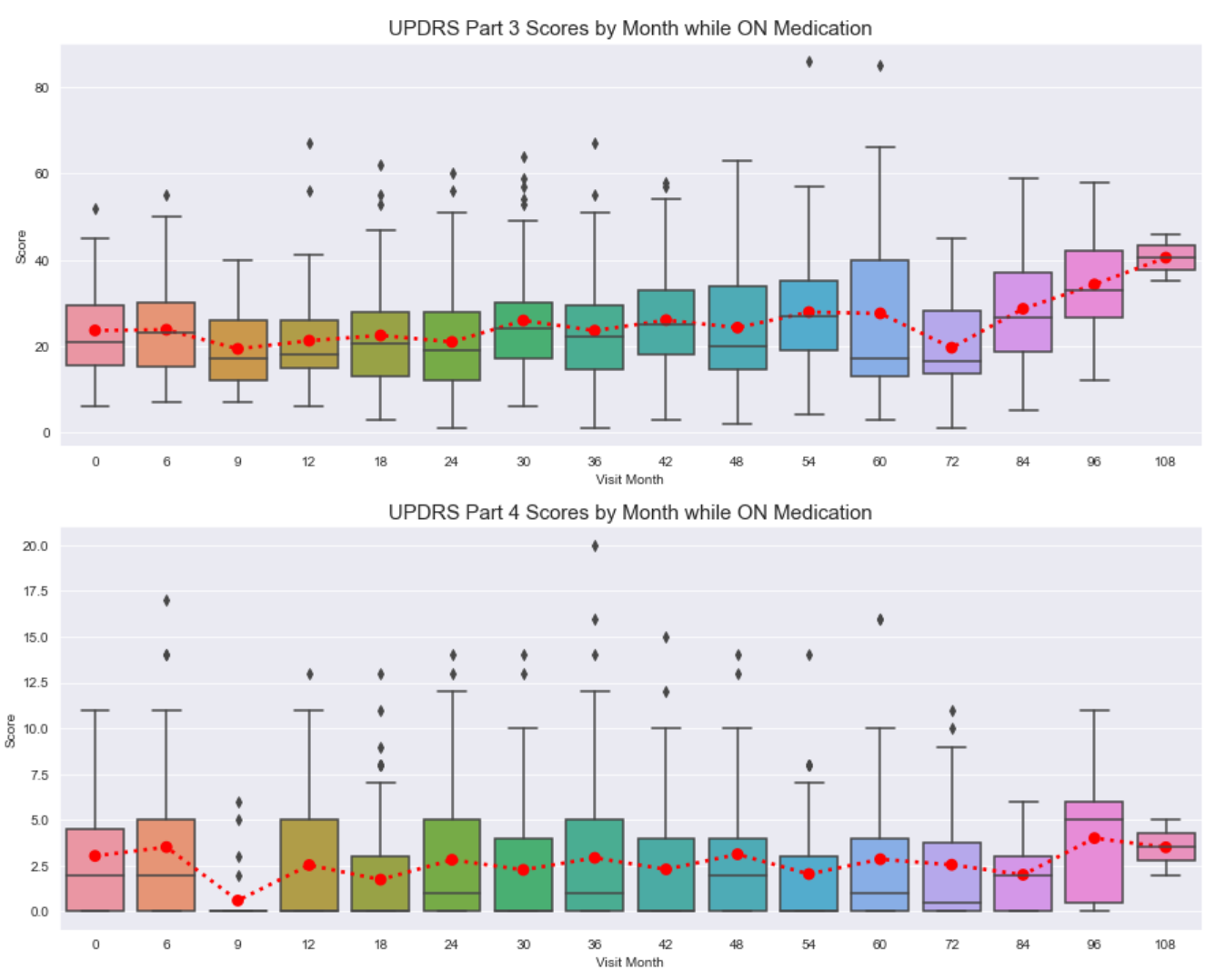
服药时的一些一般观察:
- 每个 UPDRS 部分及其各自的访问月份之间存在大量差异和异常值。
- 总的来说,在 UPDRS 第 1、2 和 4 部分中,分数的趋势线保持相对平坦。
- 对于 UPDRS 第 3 部分,我们看到分数逐渐增加。
正如 Holden 等人 (2018) 所提到的,UPDRS 的最高得分为 272。在 UPDRS 的先前版本中,UPDRS 得分随着时间的推移呈线性增长。 我们应该查看 UPDRS 分数的总和,看看该数据中的分数是否随时间增加。
train_clinical_data["updrs_sum"] = train_clinical_data["updrs_1"] + train_clinical_data["updrs_2"] + train_clinical_data["updrs_3"] + train_clinical_data["updrs_4"]
train_clincial_data_copy = train_clinical_data.copy()
train_clincial_data_copy["upd23b_clinical_state_on_medication"] = train_clincial_data_copy["upd23b_clinical_state_on_medication"].fillna("Off")
fig, axs = plt.subplots(nrows=2, ncols=1, figsize=(15, 12.5))
axs = axs.flatten()
sns.set_style('darkgrid')
data = train_clincial_data_copy[(train_clincial_data_copy["upd23b_clinical_state_on_medication"] == "Off")]
ax = axs[0]
_ = sns.boxplot(data=data, x="visit_month", y="updrs_sum", ax=ax)
_ = sns.pointplot(data=data, x="visit_month", y="updrs_sum", color="r", ci=None, linestyles=[":"], ax=ax)
_ = ax.set_title("UPDRS Sum of Scores by Month while OFF Medication".format(x+1), fontsize=15)
_ = ax.set_xlabel("Visit Month")
_ = ax.set_ylabel("Score")
data = train_clincial_data_copy[(train_clincial_data_copy["upd23b_clinical_state_on_medication"] == "On")]
ax = axs[1]
_ = sns.boxplot(data=data, x="visit_month", y="updrs_sum", ax=ax)
_ = sns.pointplot(data=data, x="visit_month", y="updrs_sum", color="r", ci=None, linestyles=[":"], ax=ax)
_ = ax.set_title("UPDRS Sum of Scores by Month while ON Medication".format(x+1), fontsize=15)
_ = ax.set_xlabel("Visit Month")
_ = ax.set_ylabel("Score")
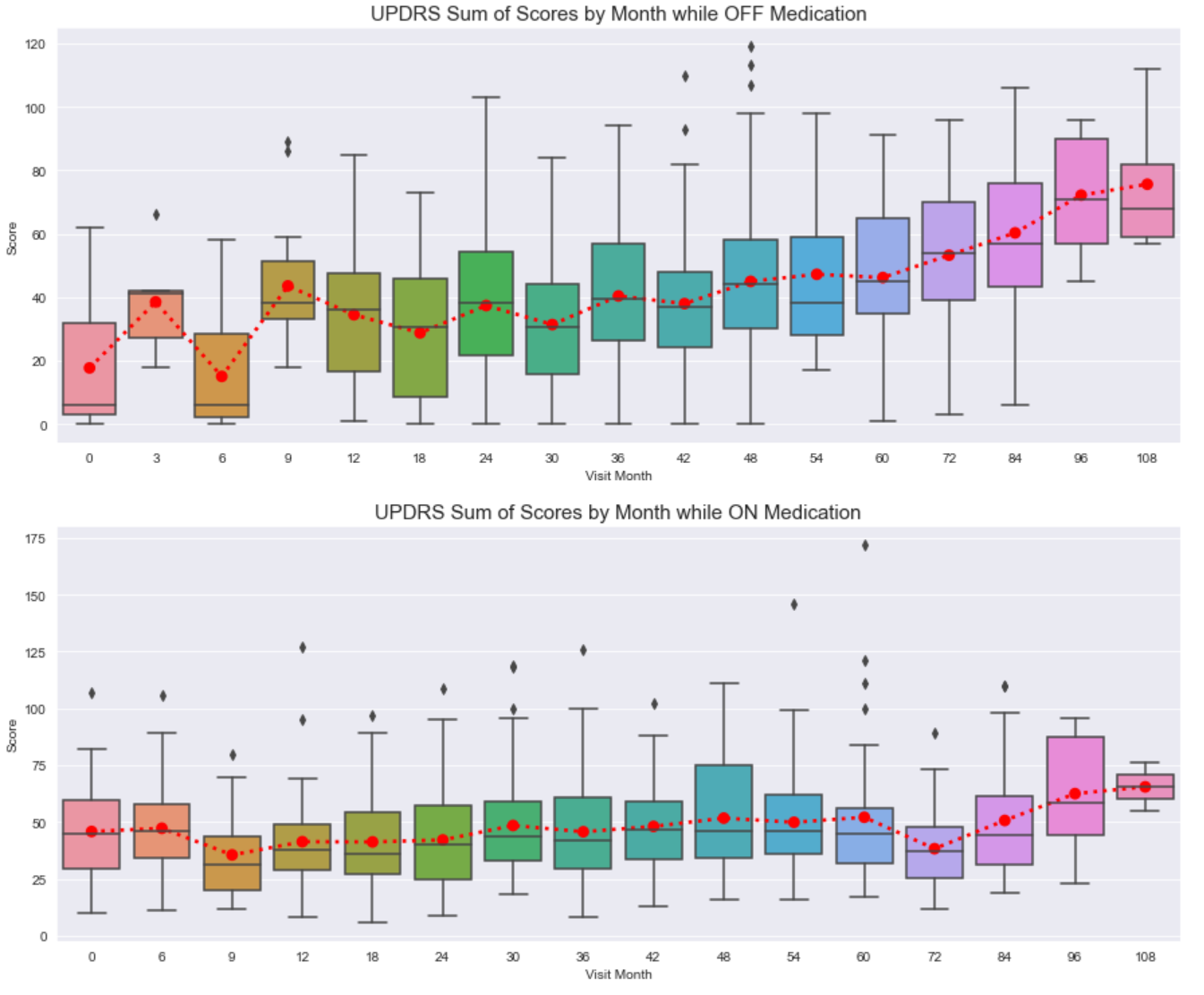
根据停药时所有 UPDRS 评分的总和,我们看到随着就诊月份的增加呈上升趋势,这表明总体而言,疾病正在进展。 在服药期间,趋势线保持相对平稳,直到月数 > 96,总分有所增加。 同样,这表明正在发生疾病进展。 如果我们结合 ON 和 OFF 药物状态:
train_clinical_data["updrs_sum"] = train_clinical_data["updrs_1"] + train_clinical_data["updrs_2"] + train_clinical_data["updrs_3"] + train_clinical_data["updrs_4"]
train_clincial_data_copy = train_clinical_data.copy()
train_clincial_data_copy["upd23b_clinical_state_on_medication"] = train_clincial_data_copy["upd23b_clinical_state_on_medication"].fillna("Off")
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(15, 5))
sns.set_style('darkgrid')
_ = sns.boxplot(data=train_clincial_data_copy, x="visit_month", y="updrs_sum", ax=ax)
_ = sns.pointplot(data=train_clincial_data_copy, x="visit_month", y="updrs_sum", color="r", ci=None, linestyles=[":"], ax=ax)
_ = ax.set_title("UPDRS Sum of Scores by Month", fontsize=15)
_ = ax.set_xlabel("Visit Month")
_ = ax.set_ylabel("Score")
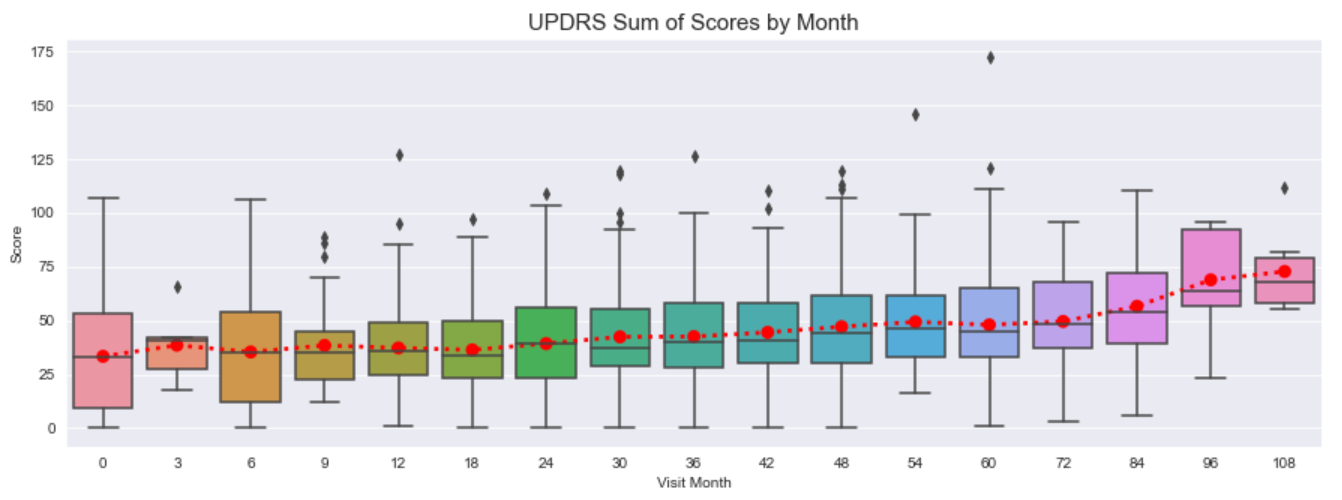
总体而言,我们看到了 UPDRS 分数增加的趋势。 这一观察很重要,因为它表明我们的分数应该会随着时间的推移而增加,而不是减少。 这可以用作后处理检查,以确保我们的机器学习算法做出的预测是有意义的。
访问月份与蛋白质数据
在不深入研究实际蛋白质数据的情况下,我们应该检查一下是否存在关于蛋白质数据按月细分的一般趋势。
train_protiens_copy = train_protiens.copy()
train_protiens_copy["log_NPX"] = np.log(train_protiens_copy["NPX"])
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(15, 5))
sns.set_style('darkgrid')
_ = sns.boxplot(data=train_protiens_copy, x="visit_month", y="log_NPX", ax=ax)
_ = sns.pointplot(data=train_protiens_copy, x="visit_month", y="log_NPX", color="r", ci=None, linestyles=[":"], ax=ax)
_ = ax.set_title("NPX by Month", fontsize=15)
_ = ax.set_xlabel("Visit Month")
_ = ax.set_ylabel("NPX")
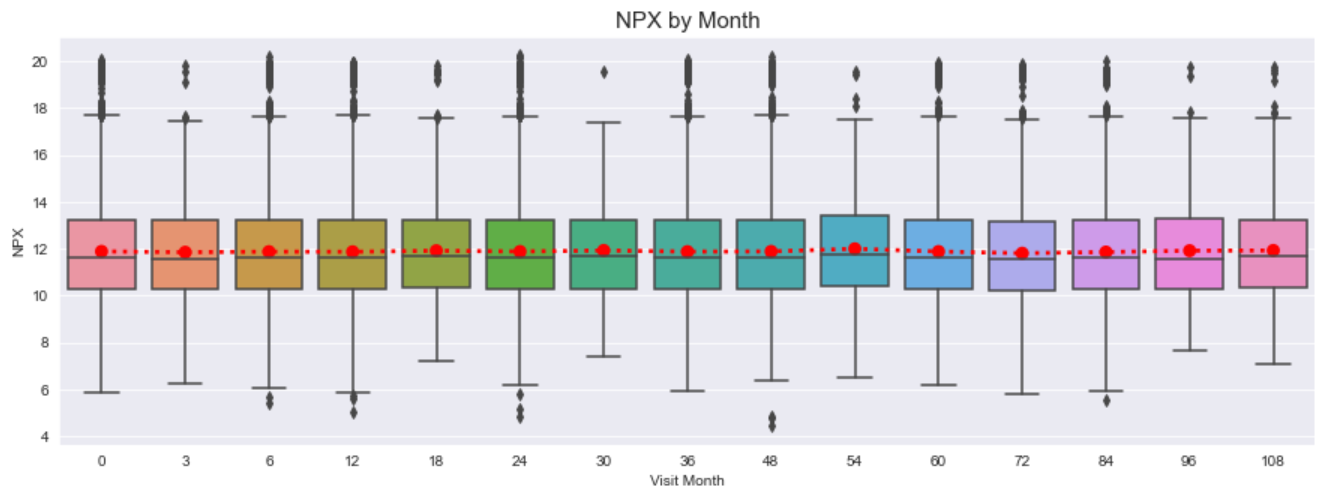
不出所料,我们在每个月类别中都看到了稳定数量的蛋白质表达。 蛋白质表达频率存在很大差异,但总的来说,我们看到几个月内重复出现相同的平均总 NPX 值。 这可能意味着实际表达的蛋白质会有差异,而不是它们的绝对数量。
我们可以检查“UniProt”蛋白质在几个月内是否有任何显着增加或减少。 查看所有 227 种蛋白质将具有挑战性。 为此,我们将研究在几个月内显着增加或减少的蛋白质。 我们将检查所有 227 种蛋白质的蛋白质计数,然后挑选出与它们的平均值相比标准偏差非常大的蛋白质。 对于此 EDA,我们将查看标准偏差超过平均值 25% 的任何蛋白质表达数据。
unique_proteins = train_protiens["UniProt"].unique()
unique_months = train_protiens["visit_month"].unique()
protein_dict = dict()
for protein in unique_proteins:
if protein not in protein_dict:
protein_dict[protein] = {
"months": unique_months,
"count_NPX": [train_protiens[(train_protiens["UniProt"] == protein) & (train_protiens["visit_month"] == month)]["NPX"].count() for month in unique_months],
"total_NPX": [train_protiens[(train_protiens["UniProt"] == protein) & (train_protiens["visit_month"] == month)]["NPX"].sum() for month in unique_months],
"avg_NPX": [0 * len(unique_months)],
}
for protein in unique_proteins:
protein_dict[protein]["avg_NPX"] = [float(total) / count for total, count in zip(protein_dict[protein]["total_NPX"], protein_dict[protein]["count_NPX"])]
for protein in unique_proteins:
protein_dict[protein]["min_NPX"] = min(protein_dict[protein]["avg_NPX"])
protein_dict[protein]["max_NPX"] = max(protein_dict[protein]["avg_NPX"])
for protein in unique_proteins:
protein_dict[protein]["mean"] = sum(protein_dict[protein]["avg_NPX"]) / len(protein_dict[protein]["months"])
protein_dict[protein]["std"] = sum([(total_NPX - protein_dict[protein]["mean"]) ** 2 for total_NPX in protein_dict[protein]["avg_NPX"]]) / (len(unique_months) - 1)
protein_dict[protein]["std"] = protein_dict[protein]["std"] ** 0.5
proteins_with_large_std = [protein for protein in unique_proteins if protein_dict[protein]["std"] > (protein_dict[protein]["mean"] * .25)]
proteins_of_interest_by_month = {
"UniProt": [],
"Visit Month": [],
"Average NPX": [],
}
for protein in proteins_with_large_std:
for month_index, month in enumerate(unique_months):
proteins_of_interest_by_month["UniProt"].append(protein)
proteins_of_interest_by_month["Visit Month"].append(month)
value = protein_dict[protein]["avg_NPX"][month_index]
value /= protein_dict[protein]["max_NPX"]
proteins_of_interest_by_month["Average NPX"].append(value)
df = pd.DataFrame(proteins_of_interest_by_month)
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(20, 8))
sns.set_style('darkgrid')
_ = sns.lineplot(data=df, x="Visit Month", y="Average NPX", hue="UniProt", style="UniProt", ax=ax)
_ = ax.set_title("Average NPX per Protein by Month", fontsize=15)
_ = ax.set_xlabel("Visit Month")
_ = ax.set_ylabel("Average NPX")
_ = plt.legend(ncol=5)
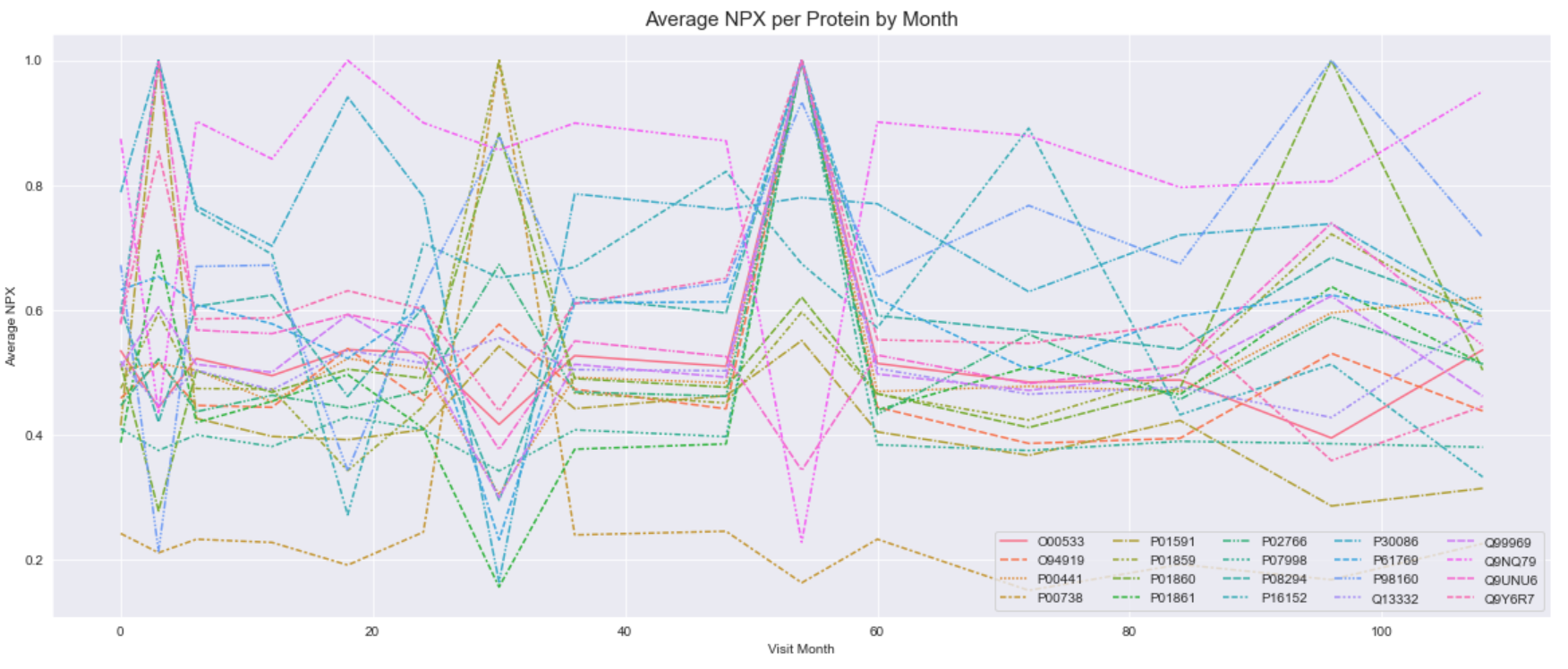
这里有一些有趣的观察结果。 首先,在第 30、54 和 96 个月,我们看到所有感兴趣的蛋白质的平均水平出现向上和向下的峰值。 这是否与药物治疗状态或 UPDRS 评分等临床数据相关,我们将在下面直接研究蛋白质特征时更详细地研究。 现在,我们只是好奇蛋白质和访问月份是否存在相关性,这显然是存在的。
第二个观察结果是几种蛋白质既有向上运动也有向下运动。 这意味着蛋白质可能与 UPDRS 评分呈正相关或负相关。
第三个也是最后一个观察结果是,我们只关注每月差异很大的蛋白质表达频率(标准差 > 平均值的 25%)。 机器学习算法可以从中学习其他更微妙的表达频率变化。
蛋白质 UniProt 和 NPX
虽然我们已经开始检查“visit_month”对各种蛋白质水平的影响,但我们也可以查看蛋白质表达水平本身,看看它们与 UPDRS 分数之间是否存在相关性。 有大量的蛋白质 (227),因此在强调有趣的相关性之前,我们将首先全面了解一下。
proteins = []
protein_dict = {}
for index, row in train_protiens.iterrows():
protein = row["UniProt"]
if protein not in protein_dict:
protein_dict[protein] = {}
proteins.append(protein)
protein_dict[protein][row["visit_id"]] = row["NPX"]
peptides = []
peptide_dict = {}
for index, row in train_peptides.iterrows():
peptide = row["Peptide"]
if peptide not in peptide_dict:
peptide_dict[peptide] = {}
peptides.append(peptide)
peptide_dict[peptide][row["visit_id"]] = row["PeptideAbundance"]
train_copy = train_clinical_data.copy()
features = []
features.extend(proteins)
# Set missing values to null so our correlation matrix won't include 0 values in the correlation calculation
train_copy[features] = train_copy[features].replace(0.0, np.nan)
features.extend(["updrs_1", "updrs_2", "updrs_3", "updrs_4"])
correlation_matrix = train_copy[features].corr(method="spearman")
from matplotlib.colors import SymLogNorm
fig, axs = plt.subplots(nrows=8, ncols=1, figsize=(20, 40))
axs = axs.flatten()
_ = sns.heatmap(
correlation_matrix.iloc[-4:,0:30],
cmap=sns.diverging_palette(230, 20, as_cmap=True),
center=0, square=True, linewidths=.1, cbar=False, ax=axs[0], annot=True,
)
_ = axs[0].set_title("Spearman Correlation Matrix", fontsize=15)
_ = sns.heatmap(
correlation_matrix.iloc[-4:,30:60],
cmap=sns.diverging_palette(230, 20, as_cmap=True),
center=0, square=True, linewidths=.1, cbar=False, ax=axs[1], annot=True,
)
_ = sns.heatmap(
correlation_matrix.iloc[-4:,60:90],
cmap=sns.diverging_palette(230, 20, as_cmap=True),
center=0, square=True, linewidths=.1, cbar=False, ax=axs[2], annot=True,
)
_ = sns.heatmap(
correlation_matrix.iloc[-4:,90:120],
cmap=sns.diverging_palette(230, 20, as_cmap=True),
center=0, square=True, linewidths=.1, cbar=False, ax=axs[3], annot=True,
)
_ = sns.heatmap(
correlation_matrix.iloc[-4:,120:150],
cmap=sns.diverging_palette(230, 20, as_cmap=True),
center=0, square=True, linewidths=.1, cbar=False, ax=axs[4], annot=True,
)
_ = sns.heatmap(
correlation_matrix.iloc[-4:,150:180],
cmap=sns.diverging_palette(230, 20, as_cmap=True),
center=0, square=True, linewidths=.1, cbar=False, ax=axs[5], annot=True,
)
_ = sns.heatmap(
correlation_matrix.iloc[-4:,180:210],
cmap=sns.diverging_palette(230, 20, as_cmap=True),
center=0, square=True, linewidths=.1, cbar=False, ax=axs[6], annot=True,
)
_ = sns.heatmap(
correlation_matrix.iloc[-4:,210:227],
cmap=sns.diverging_palette(230, 20, as_cmap=True),
center=0, square=True, linewidths=.1, cbar=False, ax=axs[7], annot=True,
)
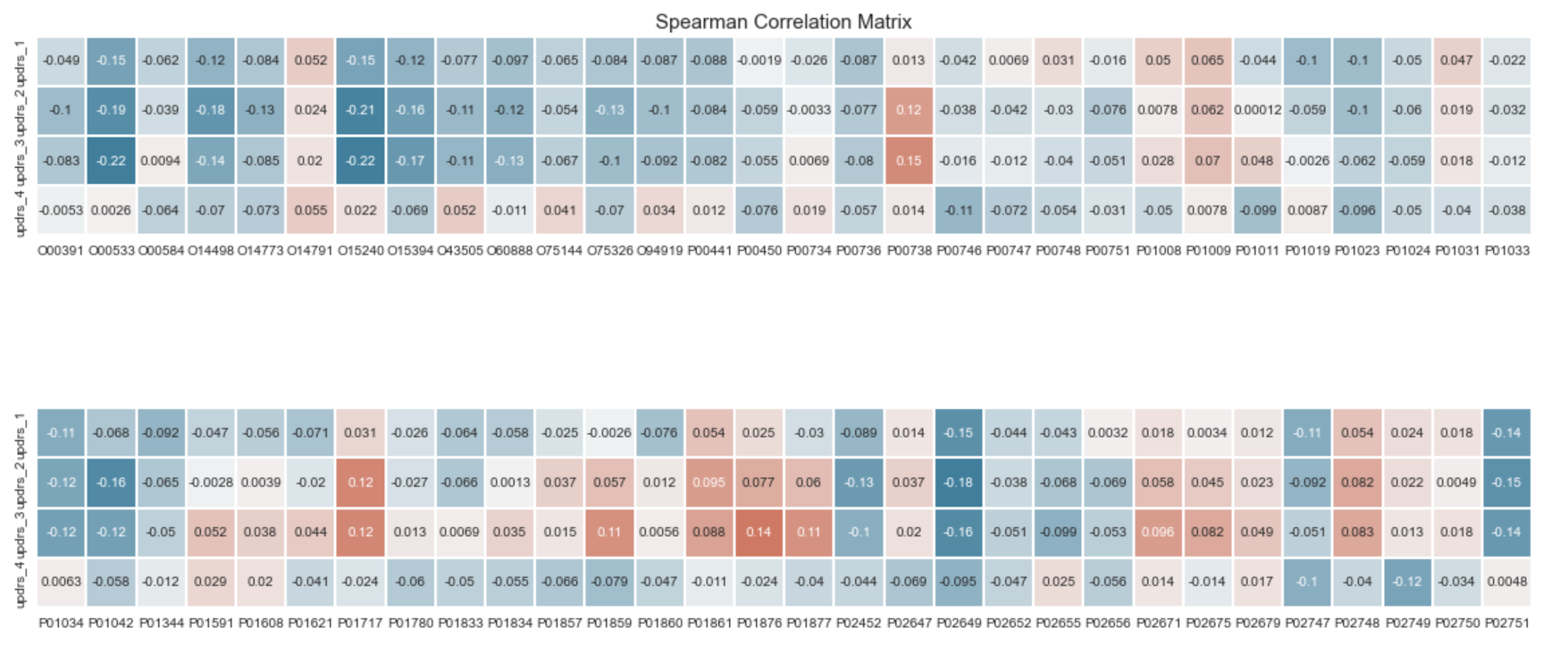

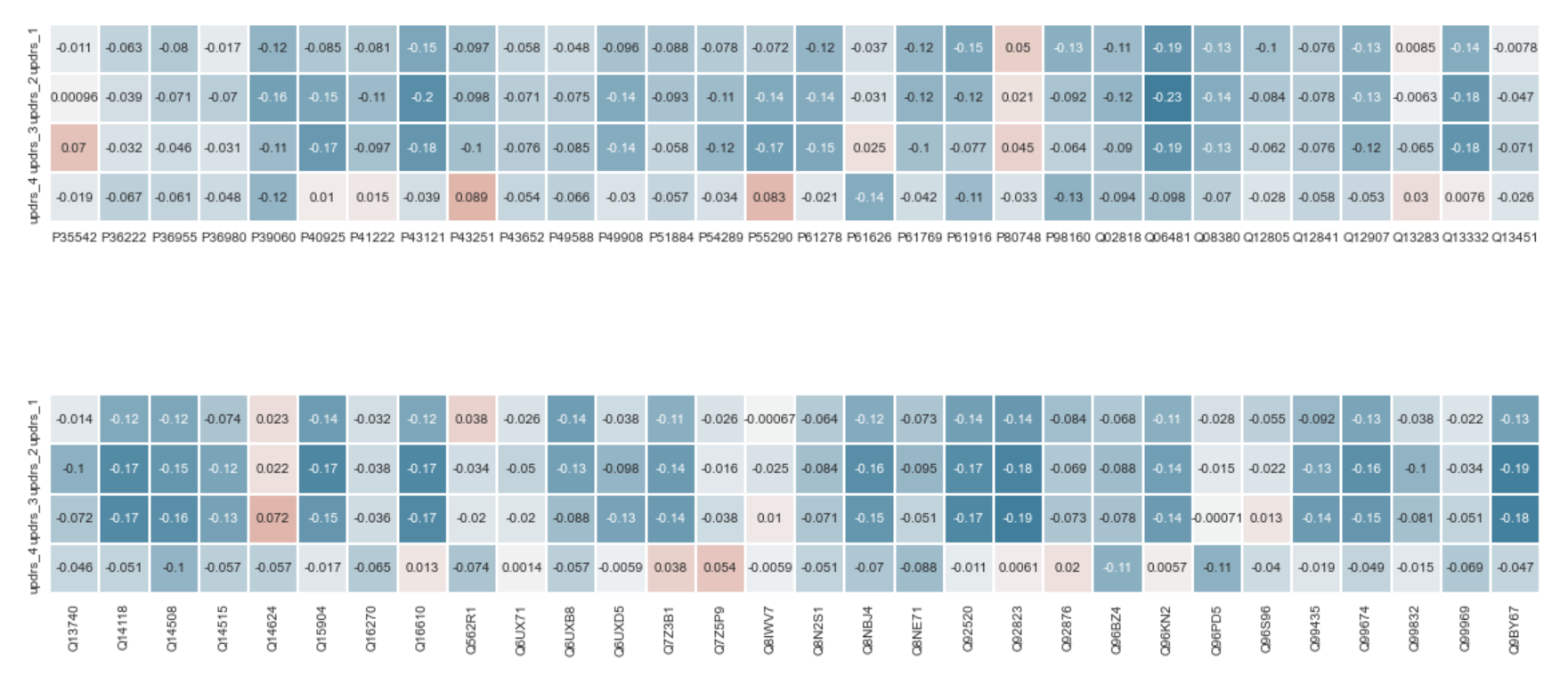
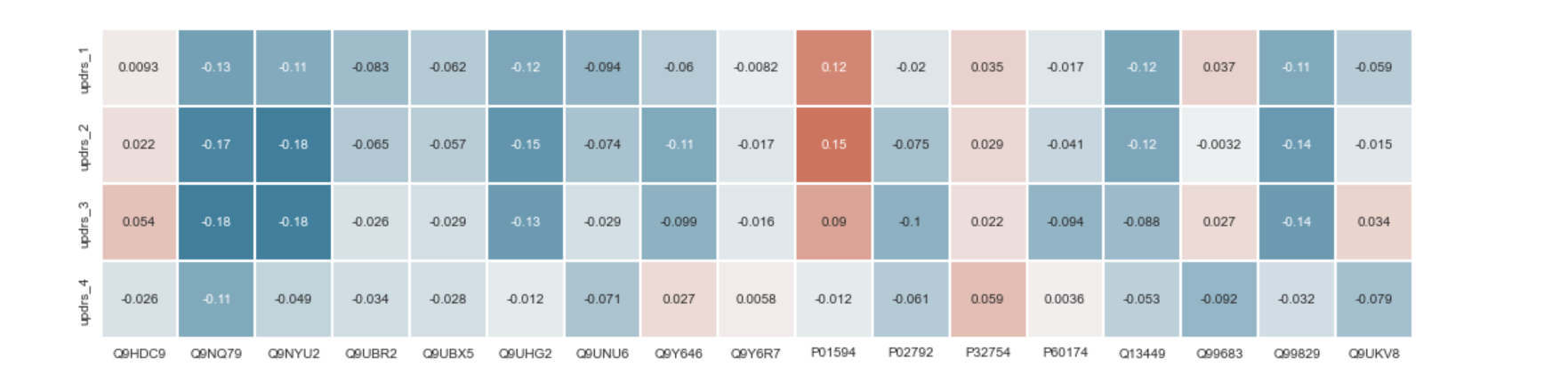
有很多蛋白质需要用相关矩阵来检查。 让我们首先定义我们认为具有某种显着相关性(正相关或负相关)的东西。 0.1 或以下的值可能与 UPDRS 目标分数几乎没有相关性,并且可能只是噪音。 快速扫描显示有几个候选者可能对我们的回归没有用:
O00533O14498O15240O15394O43505O60888P00738P01034P01042P01717P02452P02649P02751P02753P02787P04075P04156P04180P04216P05060P05067P05155P05408P05452P06396P07195P07225P07602P07711P07858P08133P08253P08571P09104P09486P09871P10645P11142P13521P13591P13611P13987P14313P14618P17174P19021P23083P23142P39060P40925P43121P49908P54289P55290P61278P61769P61916P98160Q02818Q06481Q08380Q12907Q13332Q14118Q14508Q14515Q15904Q16610Q6UXB8Q7Z3B1Q8NBJ4Q92520Q92823Q96KN2Q99435Q99674Q9BY67Q9NQ79Q9NYU2Q9UHG2P01594Q13449Q99829
有些蛋白质与 updrs_4 弱相关:
-
P00746 -
P02749 -
P02774 -
P04211 -
P04217 -
P05155 -
P06681 -
P19827 -
P20774 -
P31997 -
P61626 -
Q96BZ4 -
Q96PD5
挑战将是我们如何使用这些知识。 我们的相关性分析之所以有效,是因为我们能够忽略缺失的值。 为了让机器学习回归发挥作用,我们需要满足以下条件之一才能使用数据: -
每次访问的每种蛋白质类型都有完整的记录。
-
找出一种估算缺失值的方法。
-
使用隐式处理缺失数据的机器学习算法。
让我们来看看我们的蛋白质在访问中是如何出现的。 具体来说,我们知道有 2,615 次独立访问。 问题是在给定总访问次数的情况下,我们看到的每种蛋白质有多少。 如果我们只看到一种蛋白质三四次,即使它与 UPDRS 分数相关,也可能不会有太大帮助。
protein_counts = {}
for protein in proteins:
protein_counts[protein] = float(len(train_copy[(train_copy[protein] > 0.0)][protein])) / len(train_copy[protein]) * 100
protein_counts = dict(sorted(protein_counts.items(), key=lambda x:x[1], reverse=True))
protein_counts
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(20, 45))
_ = sns.barplot(y=list(protein_counts.keys()), x=list(protein_counts.values()), ax=ax)
_ = ax.set_title("% of Visits Containing Specified Protein", fontsize=15)
_ = ax.set_ylabel("Protein")
_ = ax.set_xlabel("% of Visits")
_ = ax.set_xlim([0, 100])
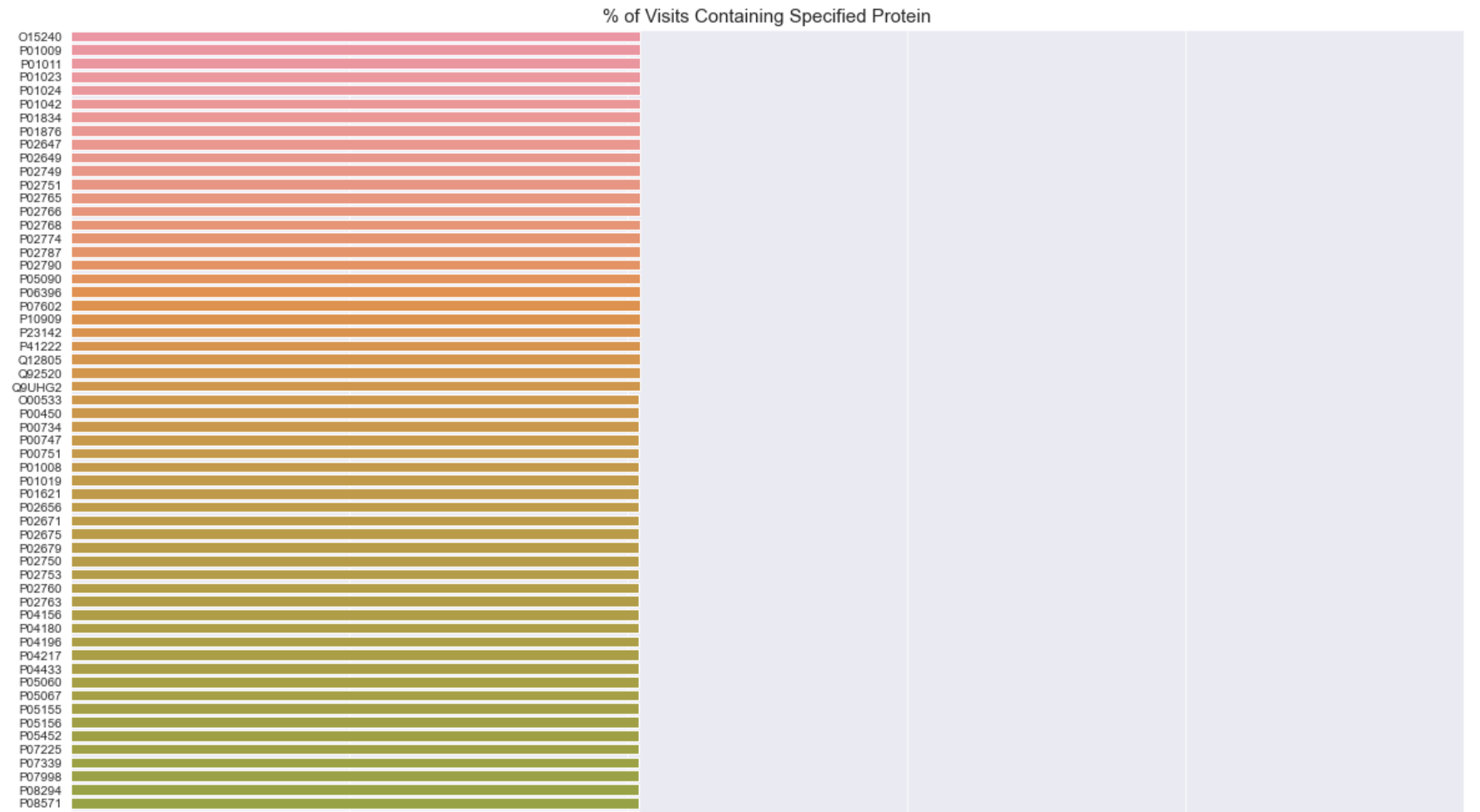


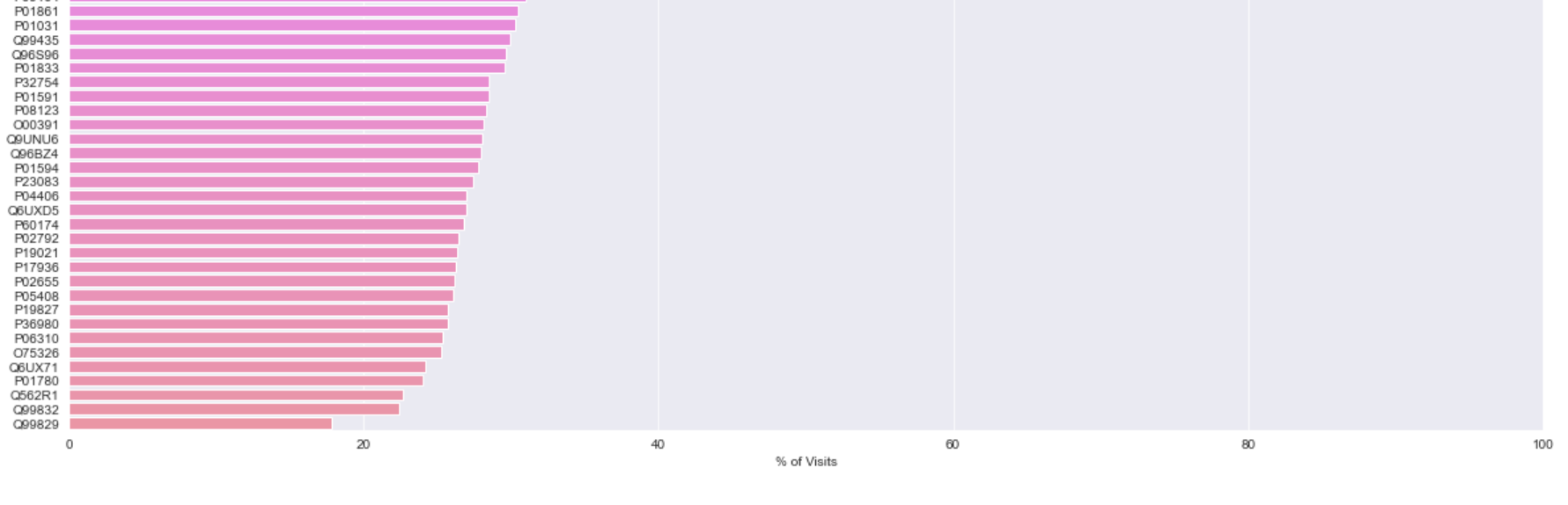
似乎对于所有蛋白质数据,蛋白质测量数据最多存在于我们记录的访问的40%。跟踪趋势会有些问题。我们没有特定蛋白质测量值的情况将压倒我们确实有蛋白质测量的例子,从而产生混淆效应。更成问题的是这些测量值来自访问月份。
protein_month_counts = {}
for protein in proteins:
protein_month_counts[protein] = {month: 0 for month in range(109)}
for x in range(109):
protein_month_counts[protein][x] = len(train_copy[(train_copy[protein] > 0.0) & (train_copy["visit_month"] == x)][protein])
from matplotlib.ticker import (MultipleLocator, AutoMinorLocator)
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(20, 5))
_ = sns.barplot(x=list(protein_month_counts["P00450"].keys()), y=list(protein_month_counts["P00450"].values()), ax=ax)
_ = ax.set_title("Samples Available by Visit Month for Protein P00450", fontsize=15)
_ = ax.set_ylabel("# Samples")
_ = ax.set_xlabel("Visit Month")
_ = ax.xaxis.set_major_locator(MultipleLocator(6))
_ = ax.xaxis.set_major_formatter('{x:.0f}')
_ = ax.xaxis.set_minor_locator(MultipleLocator(3))
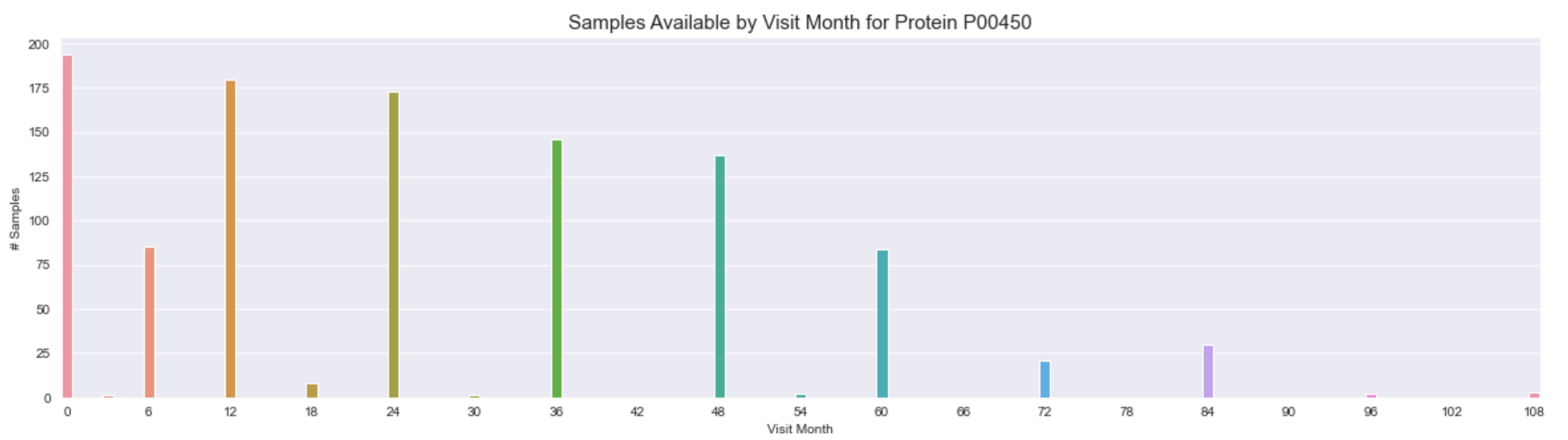
import warnings # Python 通过调用 warnings 模块中定义的 warn () 函数来发出警告
warnings.filterwarnings('ignore') # 警告过滤器
train_copy = train_copy.fillna(0)
train_copy["missing_all"] = train_copy[proteins].apply(lambda x: 1 if sum([y for y in x]) == 0 else 0, axis=1)
missing_month_counts = [train_copy[(train_copy["visit_month"] == x)]["missing_all"].sum() / float(train_copy[(train_copy["visit_month"] == x)]["patient_id"].count()) * 100 for x in range(109)]
missing_month_labels = [x for x in range(109)]
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(20, 5))
_ = sns.barplot(x=missing_month_labels, y=missing_month_counts, ax=ax)
_ = ax.set_title("% of Patients Missing Protein Data by Visit Month", fontsize=15)
_ = ax.set_ylabel("% of Patients")
_ = ax.set_xlabel("Visit Month")
_ = ax.xaxis.set_major_locator(MultipleLocator(3))
_ = ax.xaxis.set_major_formatter('{x:.0f}')
_ = ax.xaxis.set_minor_locator(MultipleLocator(3))
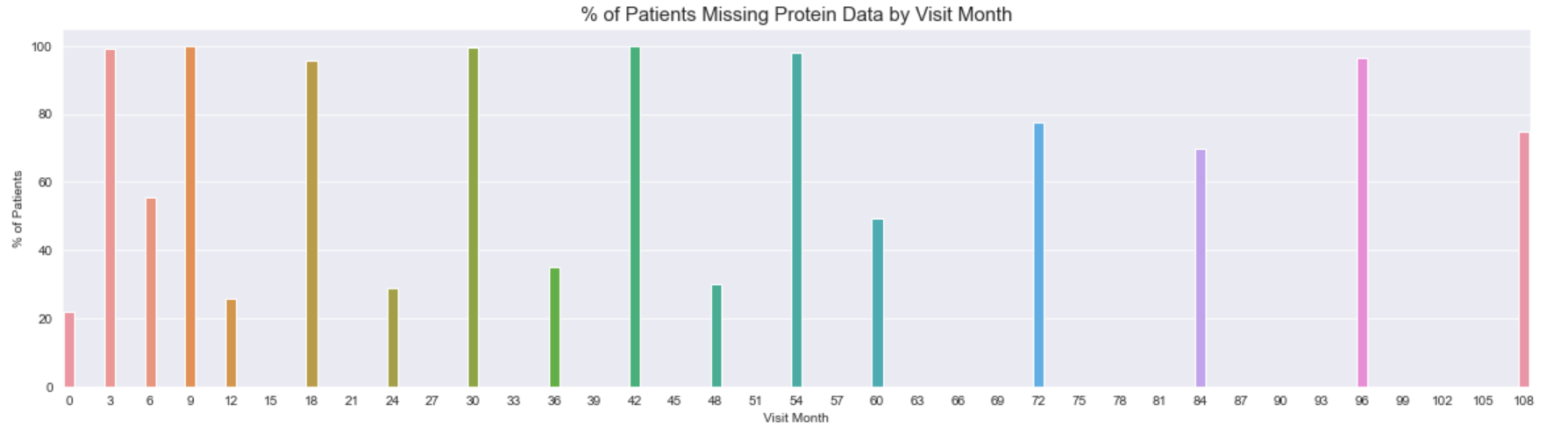
正如我们所看到的,在第3、9、18、30、42、54和96个月,我们缺乏研究中几乎所有患者的蛋白质数据。
脑脊液肽作为潜在的帕金森病生物标志物 (2015)
Shi 等人(2015 年)的研究专门研究了脑脊液蛋白和肽,它们是帕金森病的潜在指标。 在确定的那些人中,以下内容在我们可用的培训数据中可用:
- 蛋白质:
P00450- 铜蓝蛋白 (CP)P07333- 巨噬细胞集落刺激因子 1 受体 (CSF1R)P10451- 骨桥蛋白 (SPP1)P01033- 金属蛋白酶抑制剂 1 (TIMP1)P01008- 抗凝血酶-III (SERPINC1)P02647- 载脂蛋白 A-I (APOA1)P01024- 补码 C3 (C3)
*Q92876-激肽释放酶 6 (KLK6)
*肽:GAYPLSIEPIGVR- 与铜蓝蛋白 (CP) 相关EPGLC(UniMod_4)TWQSLR- 与蛋白质金属蛋白酶抑制剂 1 (TIMP1) 相关WQEEMELYR- 与蛋白质载脂蛋白 A-I (APOA1) 相关QPSSAFAAFVK- 与蛋白质补体 C3 (C3) 相关GLVSWGNIPC(UniMod_4)GSK- 与蛋白质 Kallikrein-6 (KLK6) 相关
我们应该检查这些蛋白质水平如何影响 UPDRS 分数。 让我们首先检查这些肽和蛋白质水平与各种 UPDRS 分数的相关性。
proteins = ["P00450", "P07333", "P10451", "P01033", "P01008", "P02647", "P01024", "Q92876"] # 蛋白质
peptides = ["GAYPLSIEPIGVR", "EPGLC(UniMod_4)TWQSLR", "WQEEMELYR", "QPSSAFAAFVK", "GLVSWGNIPC(UniMod_4)GSK"] # 肽
protein_dict = {}
for index, row in train_protiens.iterrows():
protein = row["UniProt"]
if protein not in protein_dict:
protein_dict[protein] = {}
protein_dict[protein][row["visit_id"]] = row["NPX"]
peptide_dict = {}
for index, row in train_peptides.iterrows():
peptide = row["Peptide"]
if peptide not in peptide_dict:
peptide_dict[peptide] = {}
peptide_dict[peptide][row["visit_id"]] = row["PeptideAbundance"]
train_copy = train_clinical_data.copy()
for protein in proteins:
train_copy[protein] = train_copy["visit_id"].apply(lambda visit_id: 0 if visit_id not in protein_dict[protein] else protein_dict[protein][visit_id])
for peptide in peptides:
train_copy[peptide] = train_copy["visit_id"].apply(lambda visit_id: 0 if visit_id not in peptide_dict[peptide] else peptide_dict[peptide][visit_id])
features = []
features.extend(proteins)
features.extend(peptides)
# Set missing values to null so our correlation matrix won't include 0 values in the correlation calculation
train_copy[features] = train_copy[features].replace(0.0, np.nan)
features.extend(["updrs_1", "updrs_2", "updrs_3", "updrs_4"])
correlation_matrix = train_copy[features].corr(method="spearman")
from matplotlib.colors import SymLogNorm
f, ax = plt.subplots(figsize=(20, 20))
_ = sns.heatmap(
correlation_matrix.iloc[13:17,0:13],
cmap=sns.diverging_palette(230, 20, as_cmap=True),
center=0,
square=True,
linewidths=.1,
cbar=False,
ax=ax,
annot=True,
)
_ = ax.set_title("Spearman Correlation Matrix for Peptides and Protiens of Interest vs UPDRS scores", fontsize=15)
# 感兴趣的肽和蛋白与 UPDRS 分数的斯皮尔曼相关矩阵
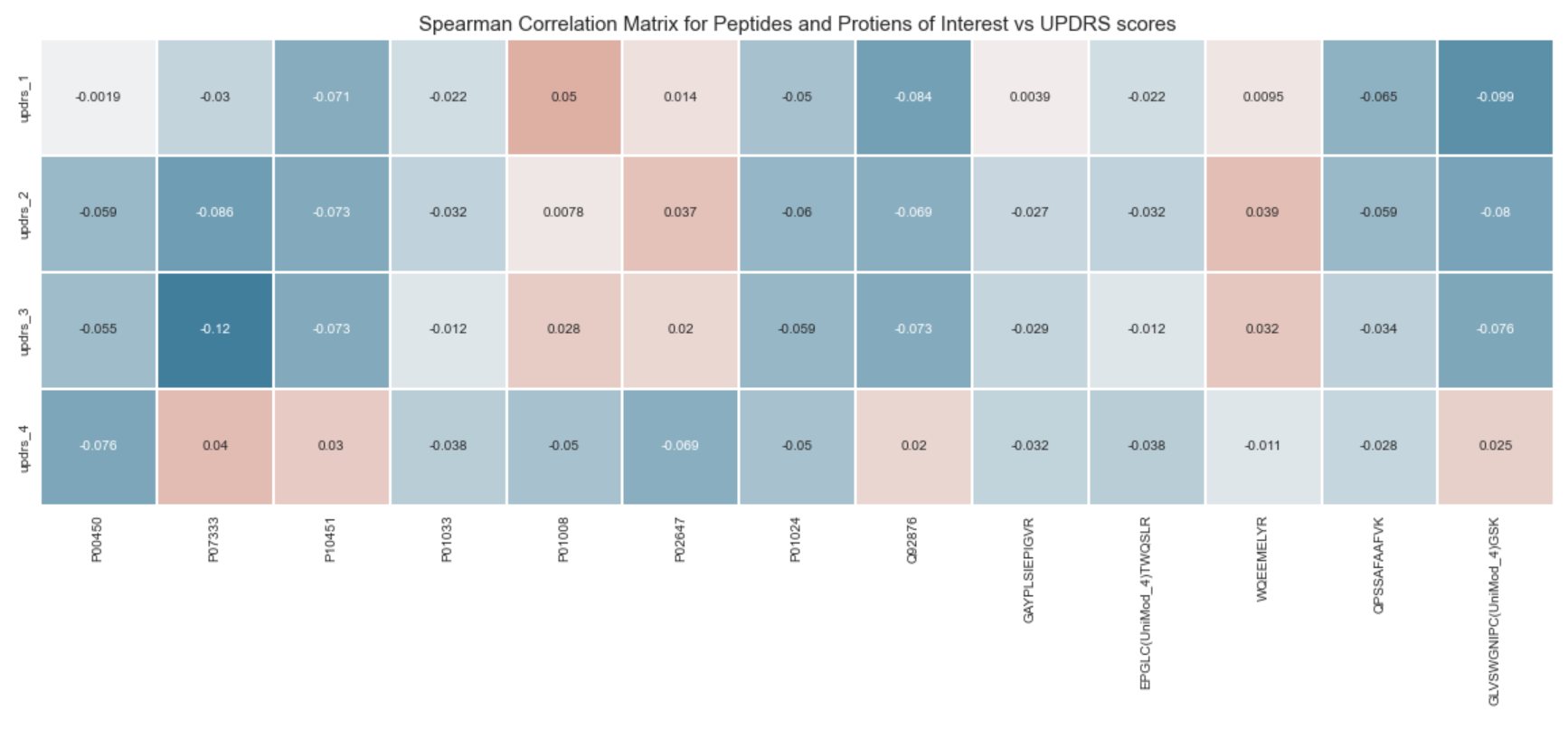
- 蛋白质
P07333与updrs_3呈弱负相关 - 肽
GLVSWGNIPC(UniMod_4)GSK与updrs_1呈弱负相关。
这些观察结果与论文中的发现一致。 简而言之,作者指出,没有任何一种单独的蛋白质或肽与 UPDRS 评分有明显的相关性——只有与其他肽和蛋白质结合时才会出现更强的信号。 该论文概述了一个与整体疾病严重程度有很强相关性的 2 肽模型。 不幸的是,训练数据集没有论文中描述的蛋白质和肽的正确组合。 尽管如此,上述两个发现可能足以提供最小的提升。
6.模型训练
我们要构建的第一个模型是一个简单的模型——我们将绝对不使用蛋白质或肽数据。 本质上,我们最终只会使用有关访问月份的先前信息来预测未来。
train_clinical_data
# 构建数据集
train_dict = {}
for index, row in train_clinical_data.iterrows():
patient_id = row["patient_id"]
visit_month = row["visit_month"]
if patient_id not in train_dict:
train_dict[patient_id] = {}
train_dict[patient_id][visit_month] = {
"updrs_1": row["updrs_1"],
"updrs_2": row["updrs_2"],
"updrs_3": row["updrs_3"],
"updrs_4": row["updrs_4"],
}
train = train_clinical_data.copy()
train["month_offset"] = 0
for index, row in train_clinical_data.iterrows():
visit_id = row["visit_id"]
patient_id = row["patient_id"]
visit_month = row["visit_month"]
month_offsets = [6, 12, 24]
for month_offset in month_offsets:
new_visit_month = visit_month + month_offset
if new_visit_month in train_dict[patient_id]:
new_row = {
"visit_id": visit_id,
"visit_month": visit_month,
"month_offset": month_offset,
"patient_id": patient_id,
"updrs_1": train_dict[patient_id][new_visit_month]["updrs_1"],
"updrs_2": train_dict[patient_id][new_visit_month]["updrs_2"],
"updrs_3": train_dict[patient_id][new_visit_month]["updrs_3"],
"updrs_4": train_dict[patient_id][new_visit_month]["updrs_4"],
"upd23b_clinical_state_on_medication": row["upd23b_clinical_state_on_medication"],
}
train = train.append(new_row, ignore_index=True)
train
from catboost import CatBoostRegressor
from sklearn.model_selection import KFold
# 0% 的 SMAPE 表示模型的预测完全准确,100% 的 SMAPE 表示模型的预测非常差,因为它低估或高估了实际值的 100%
def smape(y_true, y_pred):
denominator = (y_true + np.abs(y_pred)) / 200.0 # 分母
diff = np.abs(y_true - y_pred) / denominator
diff[denominator == 0] = 0.0
return np.mean(diff)
features = ['visit_month', 'month_offset']
train_copy = train.copy()
train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]] = train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].fillna(0)
n_folds = 5
skf = KFold(n_splits=n_folds, random_state=2023, shuffle=True)
train_oof_preds = np.zeros((train.shape[0], 4))
smape_scores = []
for fold, (train_index, test_index) in enumerate(skf.split(train_copy, train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]])):
print(f"-------> Fold {fold + 1} <--------")
x_train, x_valid = pd.DataFrame(train_copy.iloc[train_index]), pd.DataFrame(train_copy.iloc[test_index])
y_train, y_valid = train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].iloc[train_index], train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].iloc[test_index]
x_train_features = pd.DataFrame(x_train[features])
x_valid_features = pd.DataFrame(x_valid[features])
model = CatBoostRegressor(
eval_metric="MultiRMSE",
loss_function="MultiRMSE",
random_state=2023,
num_boost_round=5000,
od_type="Iter",
od_wait=200,
use_best_model=True,
verbose=0,
)
model.fit(
x_train_features[features],
y_train,
eval_set=[(x_valid_features[features], y_valid)],
verbose=0,
early_stopping_rounds=200,
use_best_model=True,
)
oof_preds = model.predict(x_valid_features[features])
train_oof_preds[test_index] = np.rint(oof_preds)
reshaped_truth = y_valid.to_numpy().reshape(-1, 1)
new_preds = np.rint(oof_preds)
reshaped_preds = new_preds.reshape(-1, 1)
local_smape = smape(reshaped_truth.flatten(), reshaped_preds.flatten())
smape_scores.append(local_smape)
print(": SMAPE = {}".format(local_smape))
smape_baseline = np.mean(smape_scores)
print("--> Overall results for out of fold predictions")
print(": SMAPE = {}".format(smape_baseline))
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(5, 5))
data = pd.DataFrame({"Fold": [x + 1 for x in range(n_folds)], "SMAPE": smape_scores})
_ = sns.lineplot(x="Fold", y="SMAPE", data=data, ax=ax)
_ = ax.set_title("SMAPE per Fold", fontsize=15)
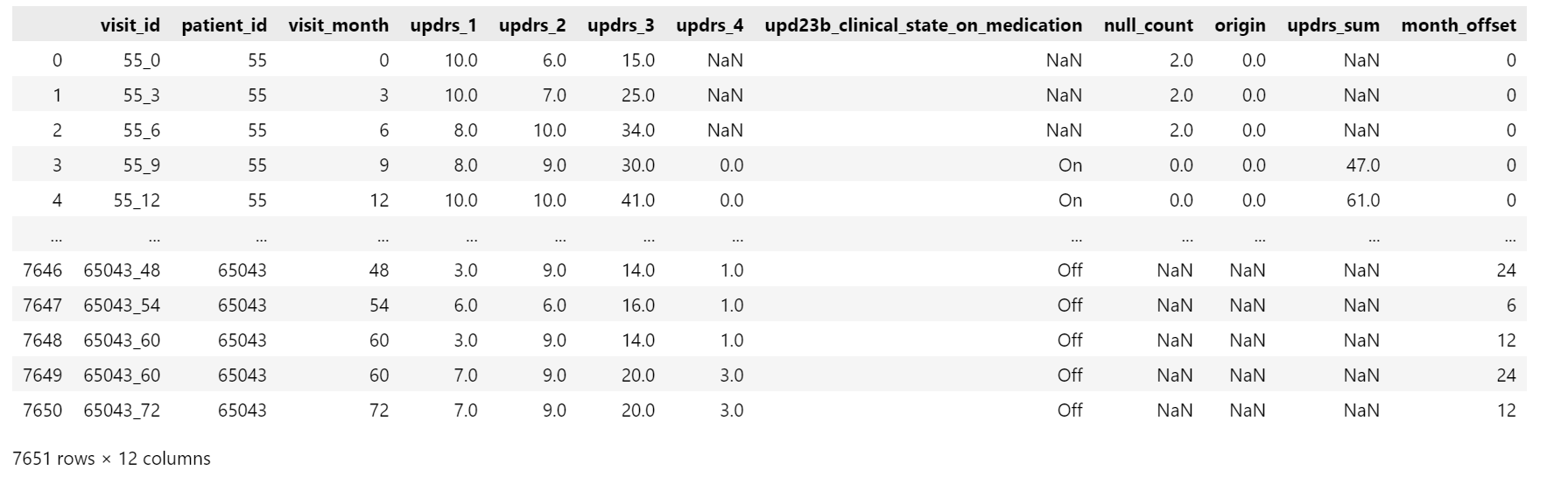
_ = ax.set_ylabel("SMAPE")
_ = ax.set_xlabel("Fold #")
正如我们所见,我们得到的 SMAPE 分数为 95.7,这不是很好。 然而,我们可以对此分数进行改进。
总的来说,由于缺失值和空值,我们的模型目前无法了解有关 UPDRS 4 的足够信息。 处理此问题的一种简单方法是将其归零。 从临床的角度来看,这不是很好,但它可能会为我们带来提升,因为我们不会多次受到惩罚,因为我们可能会预测一个应该没有的值。
features = [
'visit_month', 'month_offset',
]
train_copy = train.copy()
train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]] = train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].fillna(0)
n_folds = 5
skf = KFold(n_splits=n_folds, random_state=2023, shuffle=True)
train_oof_preds = np.zeros((train.shape[0], 4))
smape_scores = []
for fold, (train_index, test_index) in enumerate(skf.split(train_copy, train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]])):
print("-------> Fold {} <--------".format(fold + 1))
x_train, x_valid = pd.DataFrame(train_copy.iloc[train_index]), pd.DataFrame(train_copy.iloc[test_index])
y_train, y_valid = train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].iloc[train_index], train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].iloc[test_index]
x_train_features = pd.DataFrame(x_train[features])
x_valid_features = pd.DataFrame(x_valid[features])
model = CatBoostRegressor(
eval_metric="MultiRMSE",
loss_function="MultiRMSE",
random_state=2023,
num_boost_round=5000,
od_type="Iter",
od_wait=200,
use_best_model=True,
verbose=0,
)
model.fit(
x_train_features[features],
y_train,
eval_set=[(x_valid_features[features], y_valid)],
verbose=0,
early_stopping_rounds=200,
use_best_model=True,
)
oof_preds = model.predict(x_valid_features[features])
oof_preds[:, 3] = 0
train_oof_preds[test_index] = np.rint(oof_preds)
reshaped_truth = y_valid.to_numpy().reshape(-1, 1)
new_preds = np.rint(oof_preds)
reshaped_preds = new_preds.reshape(-1, 1)
local_smape = smape(reshaped_truth.flatten(), reshaped_preds.flatten())
smape_scores.append(local_smape)
print(": SMAPE = {}".format(local_smape))
smape_updrs40 = np.mean(smape_scores)
print("--> Overall results for out of fold predictions")
print(": SMAPE = {}".format(smape_updrs40))
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(5, 5))
data = pd.DataFrame({"Fold": [x + 1 for x in range(n_folds)], "SMAPE": smape_scores})
_ = sns.lineplot(x="Fold", y="SMAPE", data=data, ax=ax)
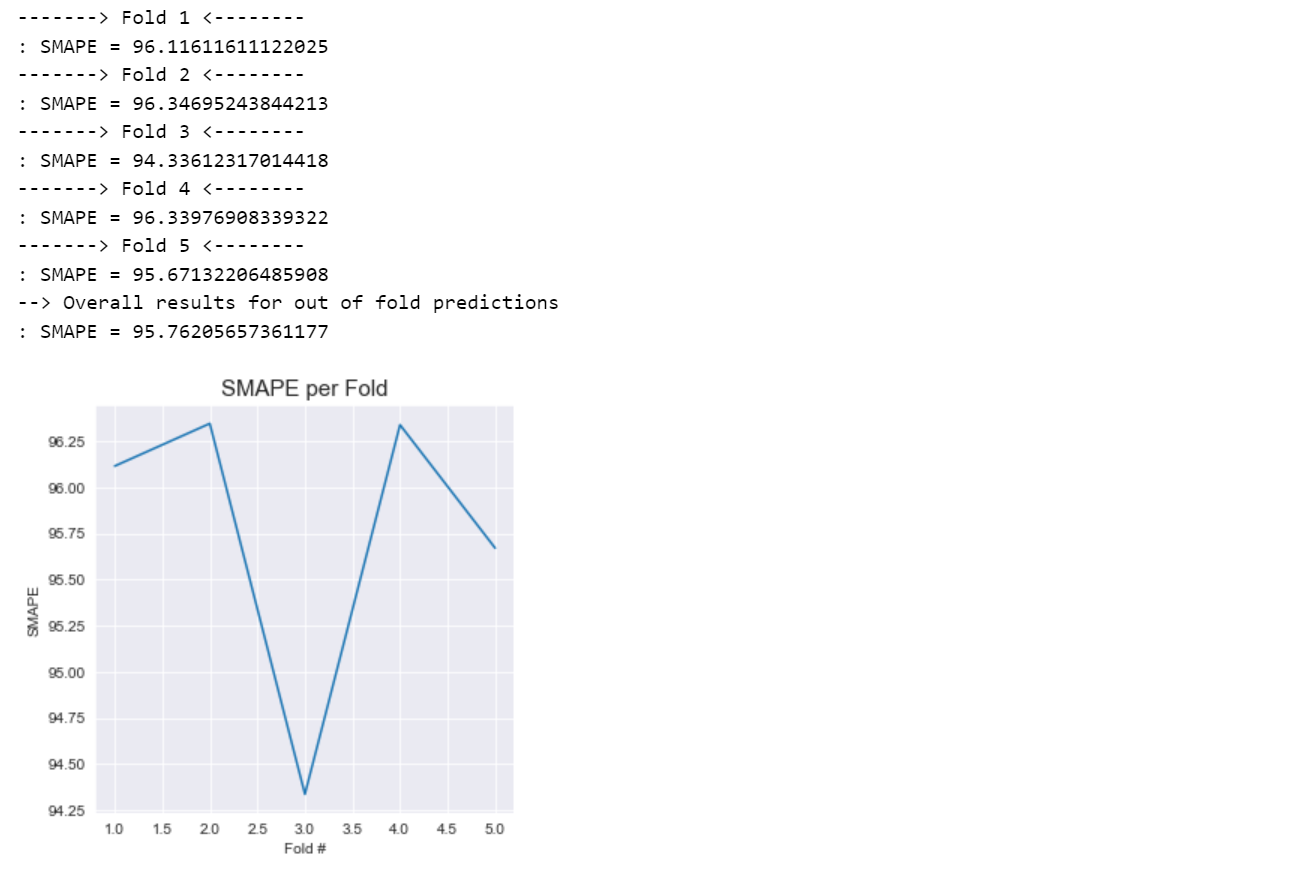
_ = ax.set_title("SMAPE per Fold", fontsize=15)
_ = ax.set_ylabel("SMAPE")
_ = ax.set_xlabel("Fold #")
正如我们所见,只需将 UPDRS 4 值设置为 0 即可提高我们的 SMAPE 分数。 这里显然有改进的空间,特别是如果我们可以利用信息做出更好的 UPDRS 4 预测。 对于未来的模型,我们将继续使用归零的 UPDRS 分数。
Supplemental Data
如果我们要继续忽略蛋白质和肽数据,那么我们可以添加额外的临床数据。 这可能会为我们带来提升,因为我们为回归器提供了更多信息以供使用。
supplemental_dict = {}
for index, row in supplemental_clinical_data.iterrows():
patient_id = row["patient_id"]
visit_month = row["visit_month"]
if patient_id not in supplemental_dict:
supplemental_dict[patient_id] = {}
supplemental_dict[patient_id][visit_month] = {
"updrs_1": row["updrs_1"],
"updrs_2": row["updrs_2"],
"updrs_3": row["updrs_3"],
"updrs_4": row["updrs_4"],
}
additional = supplemental_clinical_data.copy()
additional["month_offset"] = 0
for index, row in supplemental_clinical_data.iterrows():
visit_id = row["visit_id"]
patient_id = row["patient_id"]
visit_month = row["visit_month"]
month_offsets = [6, 12, 24]
for month_offset in month_offsets:
new_visit_month = visit_month + month_offset
if new_visit_month in supplemental_dict[patient_id]:
new_row = {
"visit_id": visit_id,
"visit_month": visit_month,
"month_offset": month_offset,
"patient_id": patient_id,
"updrs_1": supplemental_dict[patient_id][new_visit_month]["updrs_1"],
"updrs_2": supplemental_dict[patient_id][new_visit_month]["updrs_2"],
"updrs_3": supplemental_dict[patient_id][new_visit_month]["updrs_3"],
"updrs_4": supplemental_dict[patient_id][new_visit_month]["updrs_4"],
"upd23b_clinical_state_on_medication": row["upd23b_clinical_state_on_medication"],
}
additional = additional.append(new_row, ignore_index=True)
additional[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]] = additional[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].fillna(0)
features = [
'visit_month', 'month_offset',
]
train_copy = train.copy()
train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]] = train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].fillna(0)
n_folds = 5
skf = KFold(n_splits=n_folds, random_state=2023, shuffle=True)
train_oof_preds = np.zeros((train.shape[0], 4))
smape_scores = []
chunk_size = int(len(additional) / n_folds)
additional_chunks = [additional[i:i+chunk_size] for i in range(0, len(additional), chunk_size)]
for fold, (train_index, test_index) in enumerate(skf.split(train_copy, train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]])):
print("-------> Fold {} <--------".format(fold + 1))
x_train, x_valid = pd.DataFrame(train_copy.iloc[train_index]), pd.DataFrame(train_copy.iloc[test_index])
y_train, y_valid = train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].iloc[train_index], train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].iloc[test_index]
x_train_features = pd.DataFrame(x_train[features])
x_train_features = pd.concat([x_train_features, additional_chunks[fold]]).reset_index(drop=True)
y_train = y_train.append(additional_chunks[fold][["updrs_1", "updrs_2", "updrs_3", "updrs_4"]])
x_valid_features = pd.DataFrame(x_valid[features])
model = CatBoostRegressor(
eval_metric="MultiRMSE",
loss_function="MultiRMSE",
random_state=2023,
num_boost_round=5000,
od_type="Iter",
od_wait=200,
use_best_model=True,
verbose=0,
)
model.fit(
x_train_features[features],
y_train,
eval_set=[(x_valid_features[features], y_valid)],
verbose=0,
early_stopping_rounds=200,
use_best_model=True,
)
oof_preds = model.predict(x_valid_features[features])
oof_preds[:, 3] = 0
train_oof_preds[test_index] = np.rint(oof_preds)
reshaped_truth = y_valid.to_numpy().reshape(-1, 1)
new_preds = np.rint(oof_preds)
reshaped_preds = new_preds.reshape(-1, 1)
local_smape = smape(reshaped_truth.flatten(), reshaped_preds.flatten())
smape_scores.append(local_smape)
print(": SMAPE = {}".format(local_smape))
smape_additional = np.mean(smape_scores)
print("--> Overall results for out of fold predictions")
print(": SMAPE = {}".format(smape_additional))
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(5, 5))
data = pd.DataFrame({"Fold": [x + 1 for x in range(n_folds)], "SMAPE": smape_scores})
_ = sns.lineplot(x="Fold", y="SMAPE", data=data, ax=ax)
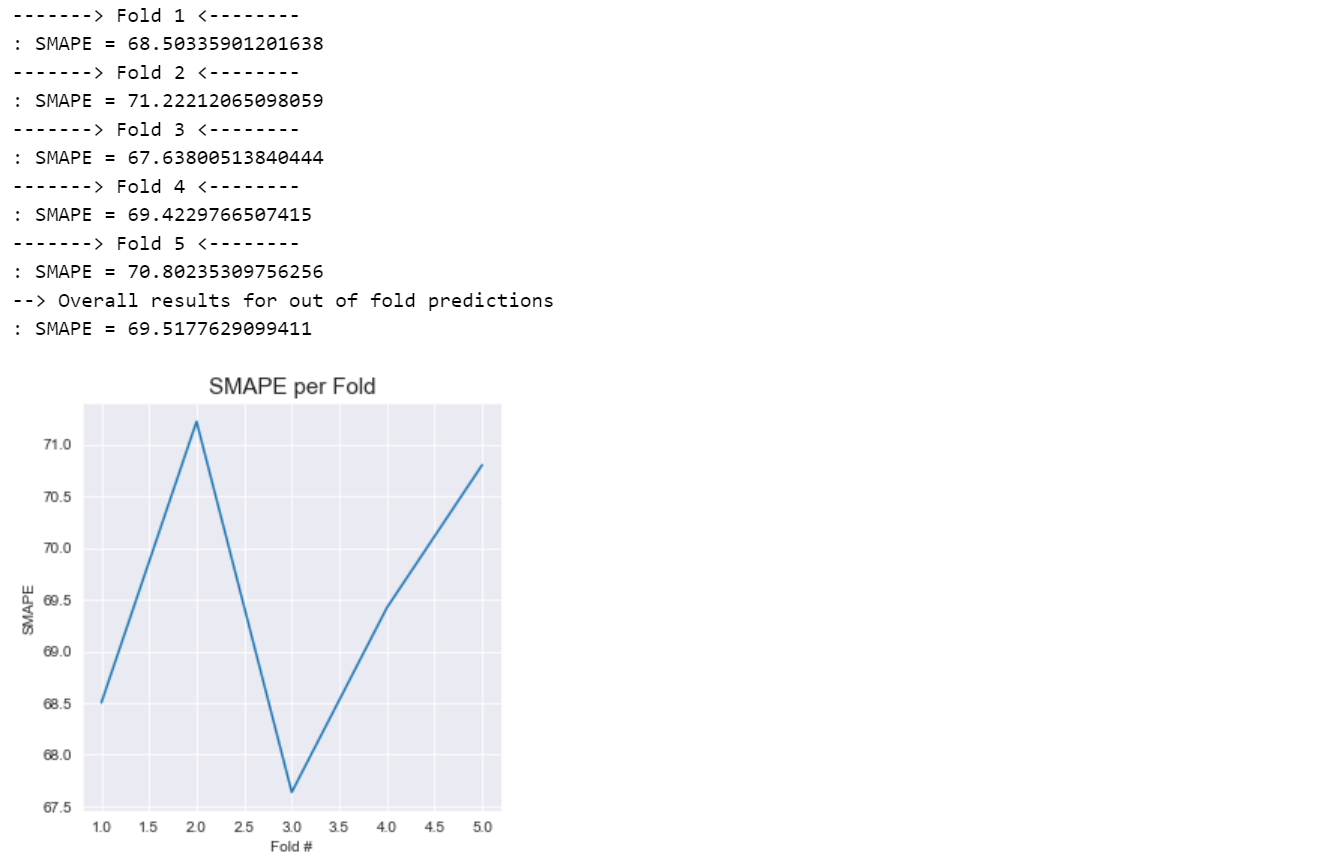
_ = ax.set_title("SMAPE per Fold", fontsize=15)
_ = ax.set_ylabel("SMAPE")
_ = ax.set_xlabel("Fold #")
与单独使用临床数据相比,我们看到了一些改进。 这可能会在隐藏测试数据不包含蛋白质信息的情况下帮助我们。
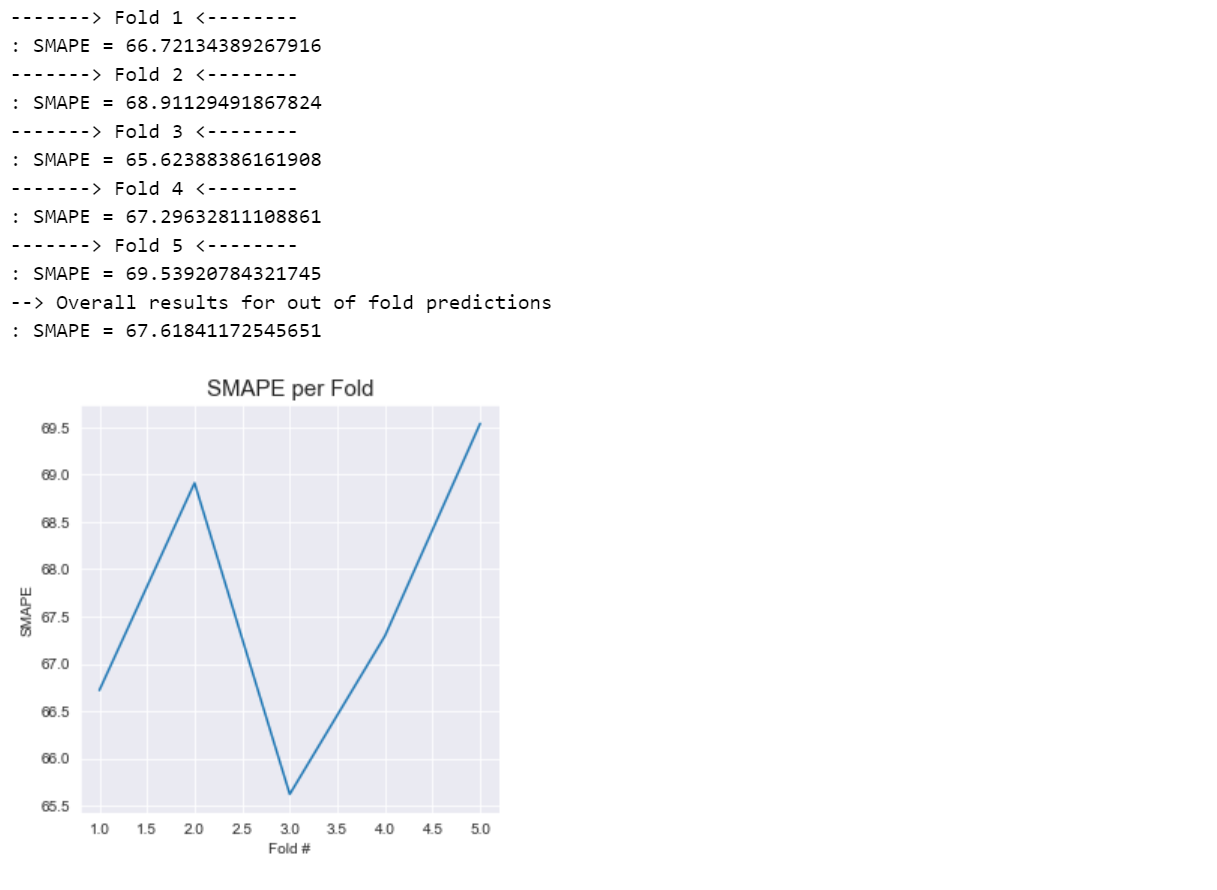
用药状态
虽然我们的测试数据中没有患者的药物治疗状态,但人们可能仍然对它感兴趣,因为我们看到药物治疗状态确实会随着时间的推移对 UPDRS 分数产生直接影响。 让我们看一下,如果我们有它,它是否有可能提供提升。
features = [
'visit_month', 'month_offset', 'med_state',
]
train_copy = train.copy()
train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]] = train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].fillna(0)
n_folds = 5
skf = KFold(n_splits=n_folds, random_state=2023, shuffle=True)
train_oof_preds = np.zeros((train.shape[0], 4))
smape_scores = []
train_copy["med_state"] = train_copy["upd23b_clinical_state_on_medication"]
train_copy["med_state"] = train_copy["med_state"].apply(lambda x: 0 if x == "Off" else 1 if x == "On" else 2)
for fold, (train_index, test_index) in enumerate(skf.split(train_copy, train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]])):
print("-------> Fold {} <--------".format(fold + 1))
x_train, x_valid = pd.DataFrame(train_copy.iloc[train_index]), pd.DataFrame(train_copy.iloc[test_index])
y_train, y_valid = train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].iloc[train_index], train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].iloc[test_index]
x_train_features = pd.DataFrame(x_train[features])
x_valid_features = pd.DataFrame(x_valid[features])
model = CatBoostRegressor(
eval_metric="MultiRMSE",
loss_function="MultiRMSE",
random_state=2023,
num_boost_round=5000,
od_type="Iter",
od_wait=200,
use_best_model=True,
verbose=0,
)
model.fit(
x_train_features[features],
y_train,
eval_set=[(x_valid_features[features], y_valid)],
verbose=0,
early_stopping_rounds=200,
use_best_model=True,
)
oof_preds = model.predict(x_valid_features[features])
oof_preds[:, 3] = 0
train_oof_preds[test_index] = np.rint(oof_preds)
reshaped_truth = y_valid.to_numpy().reshape(-1, 1)
new_preds = np.rint(oof_preds)
reshaped_preds = new_preds.reshape(-1, 1)
local_smape = smape(reshaped_truth.flatten(), reshaped_preds.flatten())
smape_scores.append(local_smape)
print(": SMAPE = {}".format(local_smape))
smape_med = np.mean(smape_scores)
print("--> Overall results for out of fold predictions")
print(": SMAPE = {}".format(smape_med))
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(5, 5))
data = pd.DataFrame({"Fold": [x + 1 for x in range(n_folds)], "SMAPE": smape_scores})
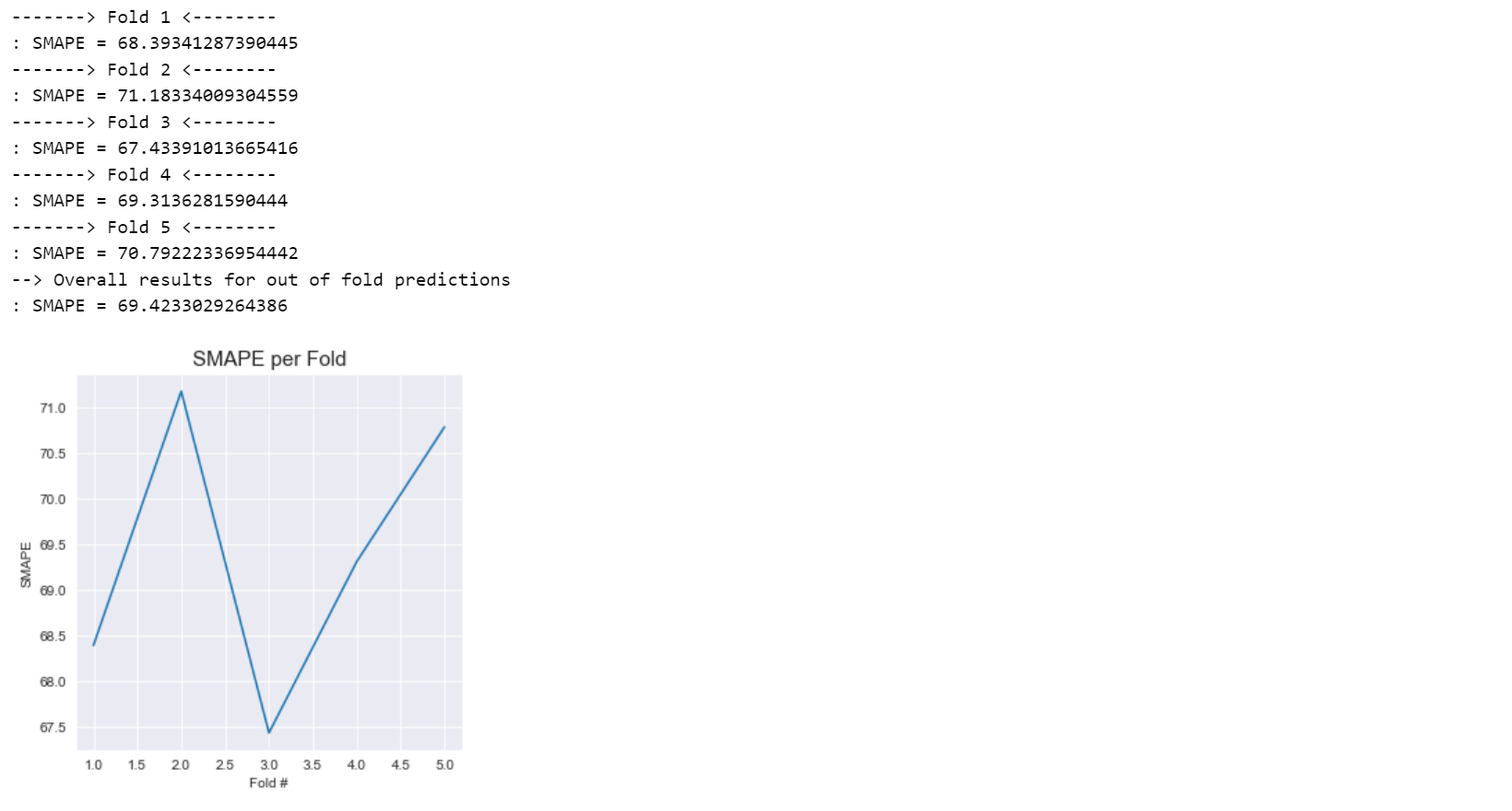
_ = sns.lineplot(x="Fold", y="SMAPE", data=data, ax=ax)
_ = ax.set_title("SMAPE per Fold", fontsize=15)
_ = ax.set_ylabel("SMAPE")
_ = ax.set_xlabel("Fold #")
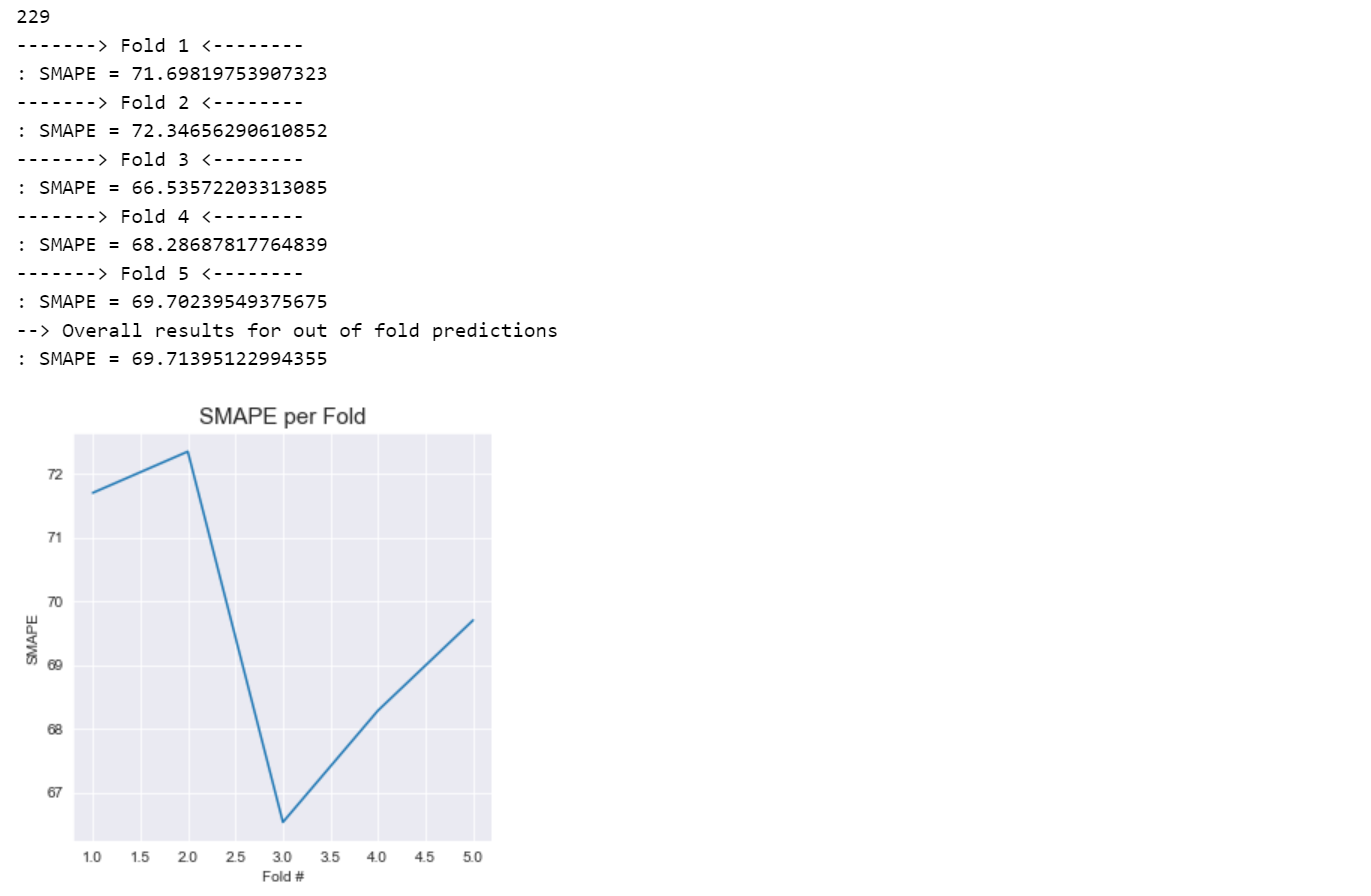
Protein Data
到目前为止,我们还没有研究过蛋白质或肽的信息。 让我们看看当我们添加时会发生什么。
from sklearn.model_selection import GroupKFold
features = [
"visit_month", "month_offset", "O00391", "O00533", "O00584", "O14498", "O14773", "O14791", "O15240",
"O15394", "O43505", "O60888", "O75144", "O75326", "O94919", "P00441", "P00450", "P00734", "P00736",
"P00738", "P00746", "P00747", "P00748", "P00751", "P01008", "P01009", "P01011", "P01019", "P01023",
"P01024", "P01031", "P01033", "P01034", "P01042", "P01344", "P01591", "P01608", "P01621", "P01717",
"P01780", "P01833", "P01834", "P01857", "P01859", "P01860", "P01861", "P01876", "P01877", "P02452",
"P02647", "P02649", "P02652", "P02655", "P02656", "P02671", "P02675", "P02679", "P02747", "P02748",
"P02749", "P02750", "P02751", "P02753", "P02760", "P02763", "P02765", "P02766", "P02768", "P02774",
"P02787", "P02790", "P04004", "P04075", "P04156", "P04180", "P04196", "P04207", "P04211", "P04216",
"P04217", "P04275", "P04406", "P04433", "P05060", "P05067", "P05090", "P05155", "P05156", "P05408",
"P05452", "P05546", "P06310", "P06396", "P06454", "P06681", "P06727", "P07195", "P07225", "P07333",
"P07339", "P07602", "P07711", "P07858", "P07998", "P08123", "P08133", "P08253", "P08294", "P08493",
"P08571", "P08603", "P08637", "P08697", "P09104", "P09486", "P09871", "P10451", "P10643", "P10645",
"P10909", "P11142", "P11277", "P12109", "P13473", "P13521", "P13591", "P13611", "P13671", "P13987",
"P14174", "P14314", "P14618", "P16035", "P16070", "P16152", "P16870", "P17174", "P17936", "P18065",
"P19021", "P19652", "P19823", "P19827", "P20774", "P20933", "P23083", "P23142", "P24592", "P25311",
"P27169", "P30086", "P31997", "P35542", "P36222", "P36955", "P36980", "P39060", "P40925", "P41222",
"P43121", "P43251", "P43652", "P49588", "P49908", "P51884", "P54289", "P55290", "P61278", "P61626",
"P61769", "P61916", "P80748", "P98160", "Q02818", "Q06481", "Q08380", "Q12805", "Q12841", "Q12907",
"Q13283", "Q13332", "Q13451", "Q13740", "Q14118", "Q14508", "Q14515", "Q14624", "Q15904", "Q16270",
"Q16610", "Q562R1", "Q6UX71", "Q6UXB8", "Q6UXD5", "Q7Z3B1", "Q7Z5P9", "Q8IWV7", "Q8N2S1", "Q8NBJ4",
"Q8NE71", "Q92520", "Q92823", "Q92876", "Q96BZ4", "Q96KN2", "Q96PD5", "Q96S96", "Q99435", "Q99674",
"Q99832", "Q99969", "Q9BY67", "Q9HDC9", "Q9NQ79", "Q9NYU2", "Q9UBR2", "Q9UBX5", "Q9UHG2", "Q9UNU6",
"Q9Y646", "Q9Y6R7", "P01594", "P02792", "P32754", "P60174", "Q13449", "Q99683", "Q99829", "Q9UKV8"
]
print(len(features))
train_copy = train.copy()
train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]] = train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].fillna(0)
proteins = train_protiens["UniProt"].unique()
protein_visit_dict = {}
for protein in proteins:
if protein not in protein_visit_dict:
protein_visit_dict[protein] = {}
for index, row in train_protiens[(train_protiens["UniProt"] == protein)].iterrows():
visit_id = row["visit_id"]
protein_visit_dict[protein][visit_id] = row["NPX"]
for protein in proteins:
train_copy[protein] = train_copy["visit_id"].apply(
lambda visit_id: protein_visit_dict[protein][visit_id] if visit_id in protein_visit_dict[protein] else 0
)
n_folds = 5
skf = GroupKFold(n_splits=n_folds)
train_oof_preds = np.zeros((train.shape[0], 4))
smape_scores = []
for fold, (train_index, test_index) in enumerate(skf.split(train_copy, train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]], groups=train_copy["patient_id"])):
print("-------> Fold {} <--------".format(fold + 1))
x_train, x_valid = pd.DataFrame(train_copy.iloc[train_index]), pd.DataFrame(train_copy.iloc[test_index])
y_train, y_valid = train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].iloc[train_index], train_copy[["updrs_1", "updrs_2", "updrs_3", "updrs_4"]].iloc[test_index]
x_train_features = pd.DataFrame(x_train[features])
x_valid_features = pd.DataFrame(x_valid[features])
model = CatBoostRegressor(
eval_metric="MultiRMSE",
loss_function="MultiRMSE",
random_state=2023,
num_boost_round=5000,
od_type="Iter",
od_wait=200,
use_best_model=True,
verbose=0,
)
model.fit(
x_train_features[features],
y_train,
eval_set=[(x_valid_features[features], y_valid)],
verbose=0,
early_stopping_rounds=200,
use_best_model=True,
)
oof_preds = model.predict(x_valid_features[features])
oof_preds[:, 3] = 0
train_oof_preds[test_index] = np.rint(oof_preds)
reshaped_truth = y_valid.to_numpy().reshape(-1, 1)
new_preds = np.rint(oof_preds)
reshaped_preds = new_preds.reshape(-1, 1)
local_smape = smape(reshaped_truth.flatten(), reshaped_preds.flatten())
smape_scores.append(local_smape)
print(": SMAPE = {}".format(local_smape))
smape_protein = np.mean(smape_scores)
print("--> Overall results for out of fold predictions")
print(": SMAPE = {}".format(smape_protein))
fig, ax = plt.subplots(nrows=1, ncols=1, figsize=(5, 5))
data = pd.DataFrame({"Fold": [x + 1 for x in range(n_folds)], "SMAPE": smape_scores})
_ = sns.lineplot(x="Fold", y="SMAPE", data=data, ax=ax)
_ = ax.set_title("SMAPE per Fold", fontsize=15)
_ = ax.set_ylabel("SMAPE")
_ = ax.set_xlabel("Fold #")
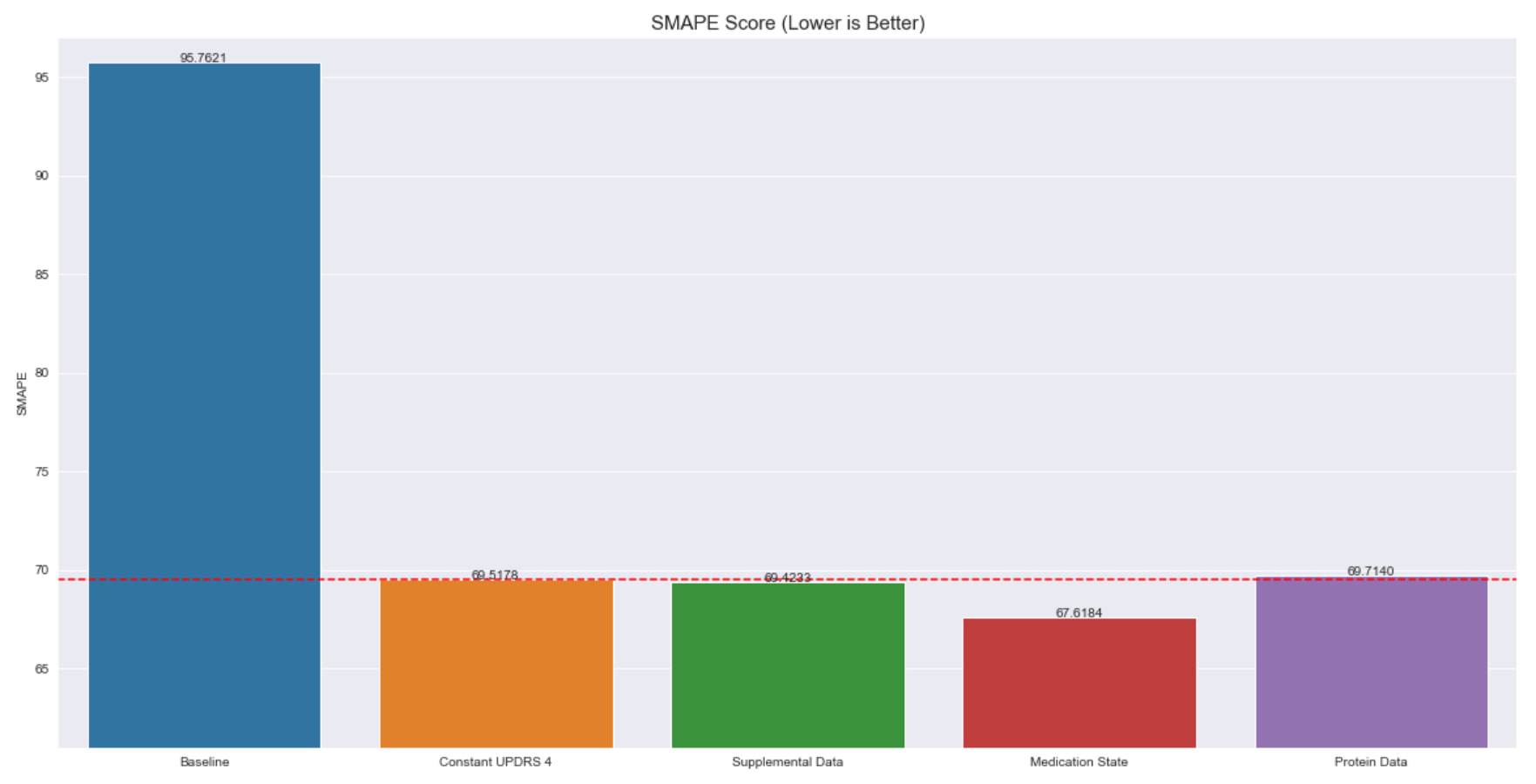
模型比较
我们可以比较每个模型的性能,看看哪个模型的性能最好。 红色虚线表示我们希望超越的基准性能。
bar, ax = plt.subplots(figsize=(20, 10))
ax = sns.barplot(
x=["Baseline", "Constant UPDRS 4", "Supplemental Data", "Medication State", "Protein Data"],
y=[
smape_baseline,
smape_updrs40,
smape_additional,
smape_med,
smape_protein,
]
)
_ = ax.axhline(y=69.51, color='r', linestyle='--')
_ = ax.set_title("SMAPE Score (Lower is Better)", fontsize=15)
_ = ax.set_xlabel("")
_ = ax.set_ylabel("SMAPE")
_ = ax.set_ylim([61, 97])
for p in ax.patches:
height = p.get_height()
ax.text(x=p.get_x()+(p.get_width()/2), y=height, s="{:.4f}".format(height), ha="center")
5.结论
根据我们的观察,有几个结论:
- 数据集的大小意味着内存压力可能不会太大。
- 临床数据中共有 248 例患者,补充数据中有 771 例患者。
- 就缺失信息而言:
- 蛋白质或肽数据中没有空条目。
- 临床数据和补充数据中有空条目。
- 我们不能假设空值表示 UPDRS 评估部件等功能的 0 值,因为 0 值表示“正常”响应。
- 患者服药状态下的空值不能假定为“开”或“关”。
- 重复数据很少出现,如果不明确过滤掉,不太可能影响机器学习模型。
- 与补充数据相比(0 - 108 个月与 0 - 36 个月),临床数据在“visit_month”中的范围似乎更大。
- 一般来说,临床数据和补充数据在数据分布方面有很大不同,正如统计和对抗性验证所证实的那样。
- 参考 Shi 等人 (2015) 的研究:
- 获得的蛋白质和肽样品不会出现在作为疾病进展或严重程度的明确指标所需的组合中。
- 我们的相关性分析表明,蛋白质“P07333”与“updrs_3”呈弱负相关,肽“GLVSWGNIPC(UniMod_4)GSK”与“updrs_1”呈弱负相关。
6.引用
- Goetz, C. G., Tilley, B. C., Shaftman, S. R., Stebbins, G. T., Fahn, S., Martinez-Martin, P., Poewe, W., Sampaio, C., Stern, M. B., Dodel, R., Dubois, B., Holloway, R., Jankovic, J., Kulisevsky, J., Lang, A. E., Lees, A., Leurgans, S., LeWitt, P. A., Nyenhuis, D., Olanow, C. W., Rascol, O., Schrag, A., Teresi, J. A., van Hilten, J. J., and LaPelle, N. (2008). Movement Disorder Society-Sponsored Revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale Presentation and Clinimetric Testing Results. Movement Disorders, 23(15), 2129–2170. DOI: 10.1002/mds.22340
- Holden, S.K., Finseth, T., Sillau, S.H. and Berman, B.D. (2018). Progression of MDS-UPDRS Scores Over Five Years in De Novo Parkinson Disease from the Parkinson’s Progression Markers Initiative Cohort. Movement Disorders Clinical Practice, 5, 47-53. DOI: 10.1002/mdc3.12553
- Shi, M., Movius, J., Dator, R. P., Aro, P., Zhao, Y., Pan, C., Lin, X., Bammler, T. K., Stewart, T., Zabetian, C. P., Peskind, E. R., Hu, S. F., Quinn, J. F., Galasko, D., & Zhang, J. (2015). Cerebrospinal Fluid Peptides as Potential Parkinson Disease Biomarkers: A Staged Pipeline for Discovery and Validation*. Molecular & Cellular Proteomics, 14(3), 544–555. DOI: 10.1074/mcp.m114.040576
笔记本地址
笔记本地址-github