摘要
SMILES是一种用于描述化合物结构的字符串表示法,其中子结构搜索是在大规模化合物数据库中查找特定的结构。然而,这种搜索方法存在一个误解,即将化合物的子结构视为一个独立的实体进行搜索,而忽略了它们在更大的化合物中的上下文。
我们使用Chemistry Development Kit (CDK)进行搜索。这种方法可以避免由于上下文缺失而导致的搜索结果不准确的问题。
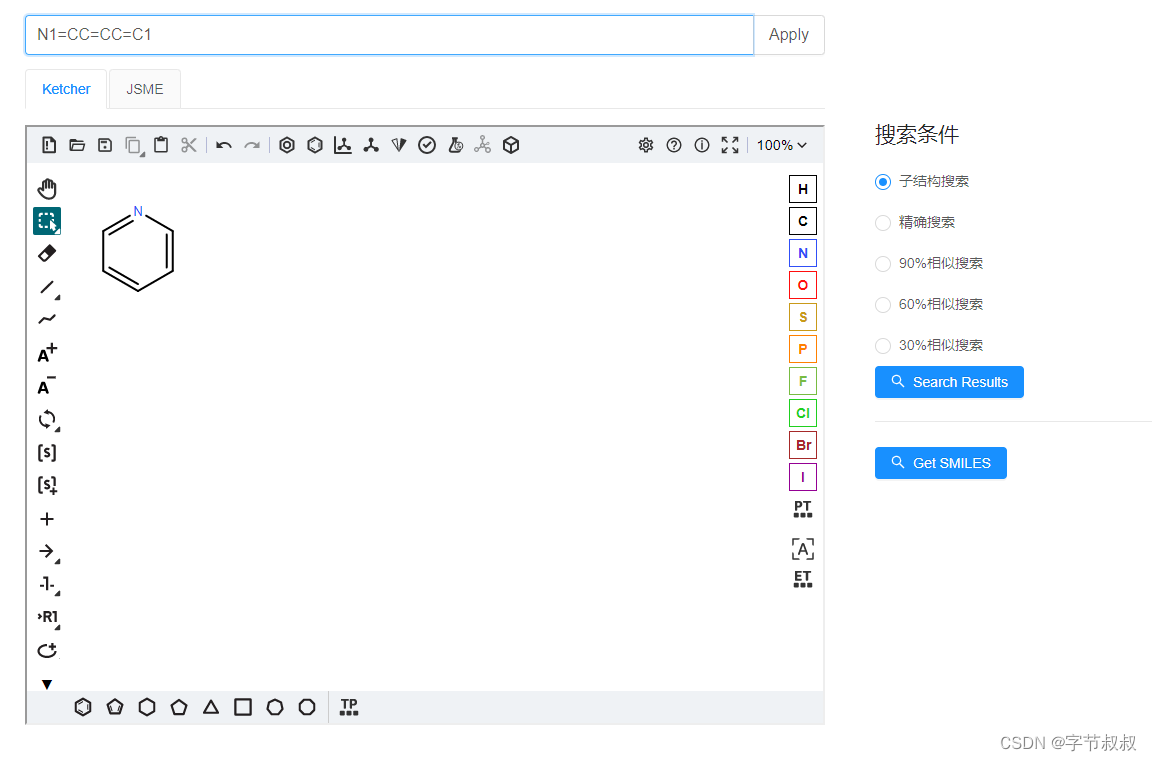
上图为我们开发的一个编辑器,用于方便查询。
Chemistry Development Kit (CDK)介绍
CDK是一种广泛使用的开源化学信息学工具包,提供表示化学概念的数据结构以及操纵此类结构和对其执行计算的方法。该库实现了各种各样的化学信息学算法,从化学结构规范化到分子描述符计算和药效团感知。它用于药物发现、代谢组学和毒理学。
为什么子串搜索≠子结构搜索
吡啶可以用 SMILES 表示:
n1ccccc1. n1ccccc1 。
但是吡啶也可以用其他五个等效的 SMILES 字符串表示:
c1ncccc1
c1cnccc1
c1ccncc1
c1cccnc1
c1ccccn1
SMILES 是从相应分子图的深度优先遍历生成的。结果,不同的起始原子和不同的相邻原子遍历将产生不同的 SMILES。一个简单的线性子串比较应该在大多数时候失败。
情况变得更糟。 SMILES 支持双键的多种编码方案。吡啶的芳香符号是一种形式。另一种形式使用大写字母和等号 ( = ) 来表示双键。六种可能性是:
N1=CC=CC=C1
C1=NC=CC=C1
C1=CN=CC=C1
C1=CC=NC=C1
C1=CC=CN=C1
C1=CC=CC=N1
情况变得更糟。单键是可选的,这意味着 N1=CC=CC=C1 等同于 N-1=C-C=C-C=C-1 。同样,芳香键是可选的,这意味着 c1ncccc1 等同于 c:1:n:c:c:c:c:1 。问题还不止于此。注意子串匹配方法在一个很好的反例中变得多么明显错误。还要考虑可以完美运行的简单案例的数量。
SMILES 解析器和生成器
IChemObjectBuilder builder = DefaultChemObjectBuilder.getInstance();
SmilesParser parse = new SmilesParser(builder);
String smiles = "N1=CC=CC=C1";
IAtomContainer mol = parse.parseSmiles(smiles);
子结构和 SMARTS 匹配
子结构匹配是基本的化学信息学操作,在指纹和描述符生成以及原子分型等许多其他功能中起着关键作用。从 CDK v1.2 开始,添加了处理 SMARTS 查询语言的功能。 SMARTS 语言得到很好的支持,包括立体化学、组件分组和原子图(以匹配反应转换)等功能。 CDK v2.0 中添加了一个新的 Pattern API,它简化了搜索结果的查找、过滤和转换。 API 是不可变的,允许一个模式被初始化一次,然后与多个分子或跨多个线程的反应进行匹配。在初始化期间,检查模式以确定需要哪些不变量(例如环大小),并且仅计算所需的不变量。内部匹配算法提供了一个惰性迭代器,以便仅在需要时才计算下一个匹配项。除了分子之外,API 还处理反应,两者都可以指定为查询或目标。
public void test2() throws InvalidSmilesException {
IChemObjectBuilder builder = DefaultChemObjectBuilder.getInstance();
SmilesParser parse = new SmilesParser(builder);
String smiles = "你的smiles表达式";
IAtomContainer mol = parse.parseSmiles(smiles);
IAtomContainer query = parse.parseSmiles("N1=C(/C=C/C(N)=O)C=CC=C1");
Pattern pattern = SmartsPattern.findSubstructure(query);
boolean matches = pattern.matches(mol);
System.out.println(matches);
}
匹配不上的情况
实践中我们发现还有一些匹配不上的情况,可能需要修改策略,可以私信我沟通。