1写在前面
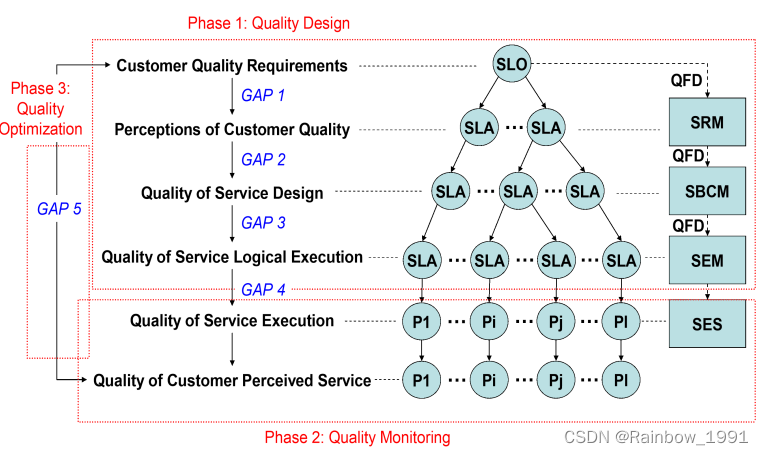
前面我们用WGCNA分析得到多个模块,其中有一些模块和我们感兴趣的表型或者临床特征是相关的。🥳
接着就是要做模块的富集分析了,帮助我们了解这些模块的基因都有哪些已知的功能,涉及到哪些通路,在哪些疾病中最为重要。🥰
现在这种做富集分析的包还是蛮多的,WGCNA包内也是内置了相关功能,不过首推的还是Y叔的clusterProfiler,在我心中真是YYDS。🫶
2用到的包
rm(list = ls())
library(WGCNA)
library(tidyverse)
library(clusterProfiler)
library(enrichplot)
library(org.Mm.eg.db)
3示例数据
load("FemaleLiver-01-dataInput.RData")
load("FemaleLiver-02-networkConstruction-auto.RData")
4整理富集分析的输入文件
一些不会使用代码的小伙伴可以采用将基因列表放到一些在线的网站上去做富集分析,如:👇
http://www.metascape.org/http://kobas.cbi.pku.edu.cn/
4.1 整理文件
annot <- read.csv(file = "./FemaleLiver-Data/GeneAnnotation.csv");
probes <- names(datExpr)
probes2annot <- match(probes, annot$substanceBXH)
allLLIDs <- annot$LocusLinkID[probes2annot];
intModules <- c("brown", "red", "salmon")
4.2 循环输出文件
这里你就可以获得你感兴趣模块的genelist了,去在线网站做富集分析吧。🤠
for (module in intModules){
modGenes <- (moduleColors==module)
modLLIDs <- allLLIDs[modGenes]
fileName <- paste("LocusLinkIDs-", module, ".txt", sep="")
write.table(as.data.frame(modLLIDs), file = fileName, row.names = F, col.names = F)
}
5内置函数进行富集分析
WGCNA包内也内置了分析函数,可以直接调用。😙
5.1 GO富集分析
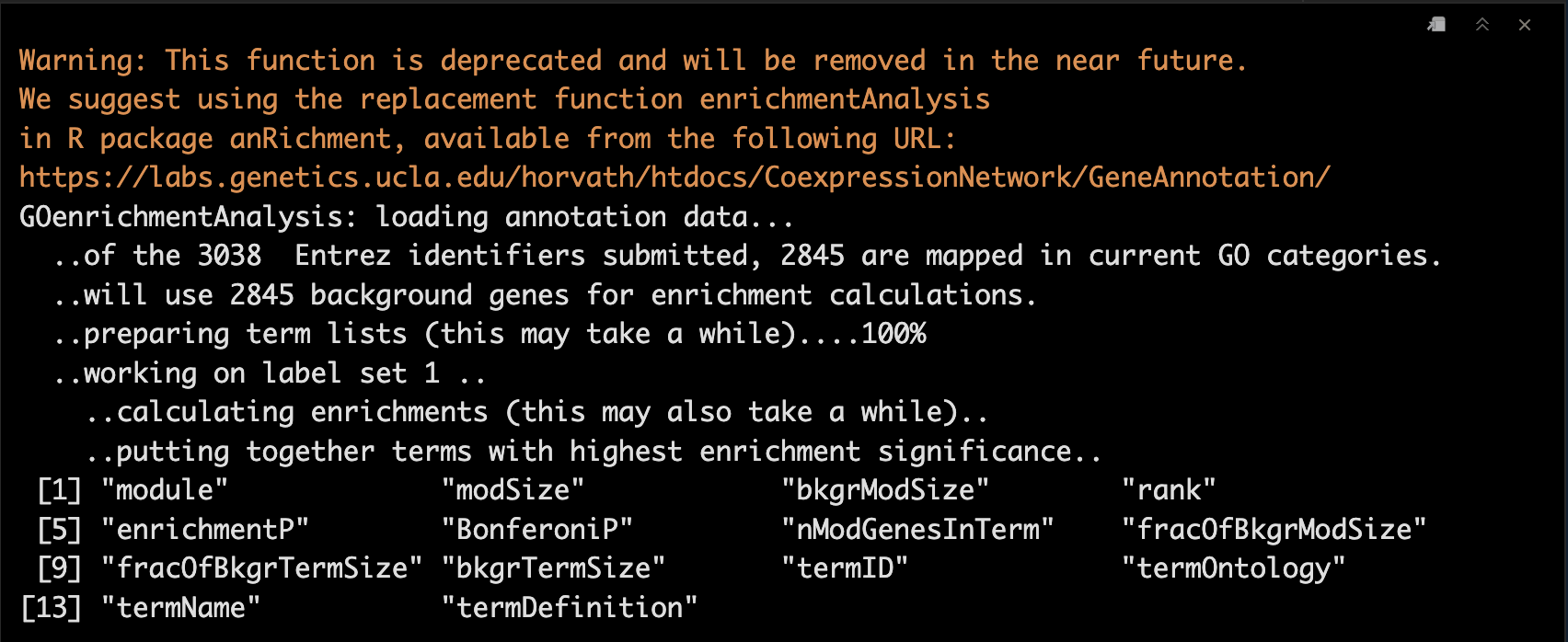
GOenr <- GOenrichmentAnalysis(moduleColors, allLLIDs, organism = "mouse", nBestP = 10);
tab <- GOenr$bestPTerms[[4]]$enrichment
names(tab)
write.table(tab, file = "GOEnrichmentTable.csv", sep = ",", quote = TRUE, row.names = FALSE)
5.2 查看结果
keepCols <- c(1, 2, 5, 6, 7, 12, 13)
screenTab <- tab[, keepCols]
numCols <- c(3, 4)
screenTab[, numCols] <- signif(apply(screenTab[, numCols], 2, as.numeric), 2)
screenTab[, 7] <- substring(screenTab[, 7], 1, 40)
colnames(screenTab) = c("module", "size", "p-val", "Bonf", "nInTerm", "ont", "term name");
rownames(screenTab) = NULL;
options(width=95)
screenTab

6clusterProfiler包进行富集分析
我们再补充一个Y叔的神包clusterProfiler包进行富集分析的方法,也是我个人最推荐的。🥰
6.1 整理输入文件
这里我们假设我们感兴趣的模块是salmon, red, brown, 我们用一下compareCluster函数,将他们都展示在一张图上。🤨
salmon <- allLLIDs[moduleColors == "salmon"] %>%
na.omit()
red <- allLLIDs[moduleColors == "red"]%>%
na.omit()
brown <- allLLIDs[moduleColors == "brown"]%>%
na.omit()
three_modules <- data.frame(id = c(salmon, red, brown),
modulecolor = rep(c("salmon", "red", "brown"),
c(length(salmon),
length(red),
length(brown))
)
)
DT::datatable(three_modules)
6.2 GO富集分析
其实不光可以做GO富集分析,KEGG, Reactome, GSEA等都是可以的,看大家自己的需求吧。🤩
formula_res.GO <- compareCluster(data = three_modules,
id~modulecolor,
fun="enrichGO",
OrgDb = 'org.Mm.eg.db')
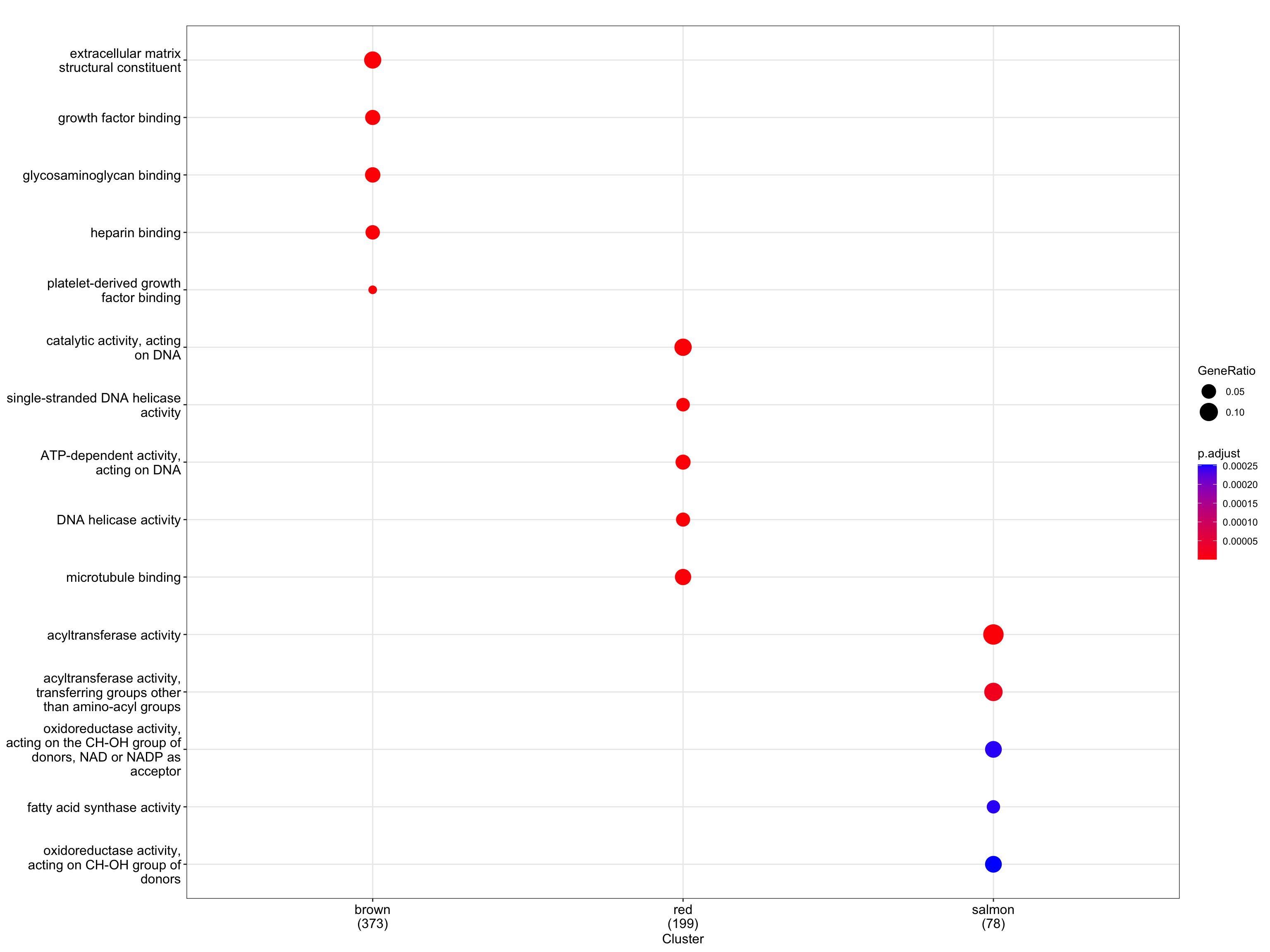
6.3 dotplot展示富集结果
Y叔写了几种可视化的方式,不做具体介绍了,大家自己喜欢什么样的就弄成什么样吧,颜色也是配成你的心头好就行。😏
dotplot(formula_res.GO)
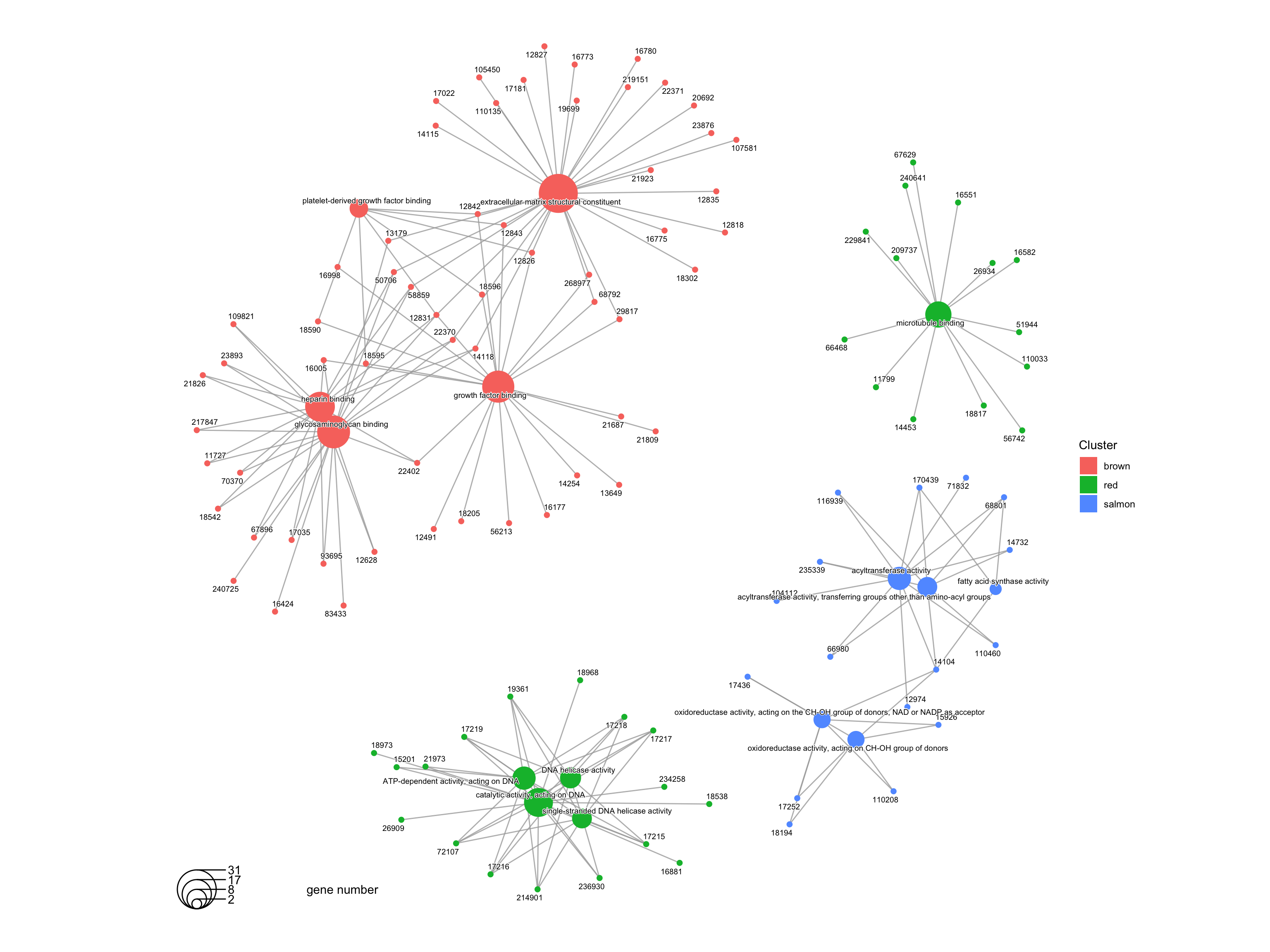
6.4 cnetplot展示富集结果
这种方式展示也不错,大家可以试试。🤪
cnetplot(formula_res.GO)
7如何引用
📍
Langfelder, P., Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). https://doi.org/10.1186/1471-2105-9-559

点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码!
📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程
📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~
📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~
📍 🧐 rms | 批量完成你的线性回归
📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图
📍 🤠 Network | 高颜值动态网络可视化工具
📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图
📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程)
📍 ......
本文由 mdnice 多平台发布