目录
- AI辅助苗头化合物筛选概述
- AI辅助CPI
- CPI数据库
- 蛋白质和化合物的特征表示
- 深度学习CPI预测
- 经典Y型架构
- 基于注意力的架构
- 基于复合物的架构
- CPI性能评估
- 苗头化合物筛选的发展前景
- 挑战与趋势
- 实际应用
AI辅助苗头化合物筛选概述
新型小分子药物的开发通常从生物学家确定疾病靶标开始,然后再成千上万的化合物中挖掘出一组可抑制或激活特定疾病靶标的活性分子。之后,再进行一系列的药代动力学,药效学,毒性测试以及结构修改获得苗头化合物(先导化合物)。接着,经过多阶段临床试验后筛选出最佳候选药。经过药监局审批,候选药物上市。
新药研发需要大量实验且具有极高失败率,完成一个新药的研发需要花费10-20年以及5-26亿美元。
高通量筛选(HTS)和虚拟筛选(VS)是获得苗头化合物的两种技术。HTS难以涵盖更多化合物,VS需要众多高质量的三维结构数据。深度学习具有强大的表征能力,可用于苗头化合物筛选中的重要步骤:化合物-蛋白质相互作用预测(CPI,compound-protrin interaction)。
AI加速CPI基于两个方面:
- 大量CPI数据可用,目前各种数据库中小分子和蛋白质之间的相互作用已经收集了数十亿条。
- 对于CPI,化合物可以表示为序列或分子图,蛋白质可以表示为序列或三维网格。这些数据结构可以被深度学习模型处理。CPI可以被视为一个图,包含两类节点:化合物和蛋白质,节点之间的边代表相互作用关系。
AI辅助CPI
CPI数据库
目前,生物实验已经积累了大量CPI数据,不仅包含小分子与蛋白质的相互作用,还包括由IC50,Ki,Kd和EC50等指标度量的结合亲和力。
STITCH是目前最大的CPI数据库,包含16亿对相互作用。BindingDB是第二大CPI数据库,包含200万个结合亲和力数据。PDBbind是中型CPI数据库,提供17000个实验确定的化合物-蛋白质复合物结构和亲和力数据,并额外包含结合位点数据。此外,KIBA,Davis,DUD-E是普遍使用的三个小型数据库。
靶点蛋白相关的数据库为KEGG,DrugBank,TTD。另外,PubChem和ChEMBL是两个综合性化合物数据库。
蛋白质和化合物的特征表示
传统的特征工程将化合物和蛋白质编码为高维特征向量,每个维度反映了化合物和蛋白质的特定属性。根据化学结构的维度,化合物的特征描述包括基于结构的1D,2D,3D表示。
另外,分子指纹是经典的化合物特征表示方法。化合物的分子指纹分为:基于子结构,基于路径,圆环,药效团。

- 化合物结构表示及其定量描述(分子指纹)
从特征工程衍生出的蛋白质描述符包括基于序列和基于结构的描述符:
- 基于序列的描述符分为基于k-order氨基酸组成和基于物理化学性质的描述符。基于k-order氨基酸组成的描述符反映了蛋白质序列中k个氨基酸组成短肽的频率。基于物理化学性质的描述符利用每个氨基酸的物理化学性质(疏水性,范德华力,极性)将氨基酸序列映射为实值序列。
- 基于结构的描述符分为基于拓扑结构,基于几何的描述符。基于拓扑结构的描述符根据从分子图生成的原子连接指数描述氨基酸。基于几何的描述符反映了与形状,大小,空间中原子位置等相关的蛋白质结构特征。
深度学习CPI预测
目前主要的方法分为三类:经典Y型,基于注意力,基于绑定复合物。
经典Y型架构
这是最早的深度学习在CPI上的应用。DeepDTA就是Y型架构,其中一个分支使用SMILES编码化合物(1D),另一个使用1D序列作为蛋白质表示编码蛋白质,然后由两个独立的卷积网络分别编码为相应的embedding。再将化合物和蛋白质embedding拼接后输入到一个或多个全连接层。最后输出亲和力预测结果。此外,化合物和蛋白质的更多特征表示也可以被整合到Y型框架。
化合物可以直接表示为分子图,因此GNN在小分子表征方面可以发挥作用。比如GraphDTA使用混合GNN(GCN,GAT,GIN图同构网络)获得化合物的图表征,采用多层1D CNN获得基于序列的蛋白质表示。
关于蛋白质,也可以通过distance map或contact map表示。比如DGraphDTA通过PconsC4从蛋白质序列生成contact map,然后构建蛋白质graph,节点为氨基酸,边表示其相邻关系,最后在小分子graph和蛋白质graph上采用GNN获得embedding。
基于注意力的架构
Y型架构实现了CPI预测,但不能指出哪些因素对相互作用有贡献,以及相应的贡献程度。将注意力整合到Y型架构有利于解释化合物与蛋白质形成的相互作用:蛋白质的关键子序列(残疾或n-gram氨基酸)与化合物的关键子结构的相互作用。
大多数基于注意力的模型分别针对化合物和蛋白质设计注意力模块。DeepCDA将化合物SMILES字符串,蛋白质序列分别传入一个LSTM和一个CNN,然后通过注意力机制表明化合物子结构和蛋白质残疾之间的相互作用强度。
一些研究设计了联合注意力模块(co-attention),AttentionDTA使用两个1D CNN提取化合物和蛋白质的表征,然后应用联合注意力模块捕获化合物子序列和蛋白质子序列,帮助寻找结合位点。
基于复合物的架构
通常,大量的蛋白质结构是难以获取的,但当有化合物-蛋白质复合物时,设计基于复合物的模型有助于CPI预测。
在早期,AtomNet直接采用3D CNN,将化合物-蛋白质复合物离散成三维网格,获得活性复合物和非活性复合物的表征。由于3D网格计算复杂,近期一些工作关注化合物-蛋白质结合口袋的特征表示(蛋白质的结合口袋是指蛋白质表面或内部具有适合与配体结合的空腔),而不是整个复合物的特征表示。

- a:Y型架构;b:结合注意力;c:基于复合物建模。
关于CPI预测
我个人认为,结合口袋建模(基于复合物的方式)可能是更合理的方法,因为基于复合物的方法从几何视角描述了化合物子结构与蛋白质子序列的相互作用。
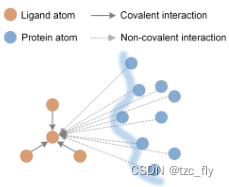
- 化合物(配体)节点从其共价和非共价邻居接收信息,其中非共价相互作用应当在CPI中主导计算。
CPI性能评估
CPI预测分为二元预测任务(分类,区分化合物是否与蛋白质结合)和亲和力预测任务(回归,推断化合物与蛋白质结合的强度)。通常,亲和力预测比二元分类预测更困难。此外,可以注意到一个集成模型DeepPourse。
- 对于二元预测,可以使用三个常用的数据集:DUD-E,Davis和Human。用AUC衡量,AUC越大,表明预测效果越好。
- 对于结合亲和力预测,通常使用Davis数据集。亲和力预测的性能使用一致性指数(Consistency index,CI)和均方误差(Mean square error,MSE)衡量,CI越大,MSE越小,预测效果越好。
苗头化合物筛选的发展前景
挑战与趋势
深度学习在CPI预测上展示了良好性能,但依然存在挑战:
- 如何利用大量未标记的化合物和蛋白质。无监督学习DeepCPI,半监督学习GANsDTA,预训练策略DeepAffinity的成功应用表明,利用丰富的未标记数据(序列)可以增强化合物和蛋白质表示。
- 由于具有标记的化合物表示数据较为稀缺,可以通过自监督学习对未标记数据预训练,然后迁移下游任务。最近工作利用自监督学习提取基于SMILES或分子图的表示。
- 虽然3D结构可以增强化合物和蛋白质的表示,但需要高额算力,而且基于复合物的模型与利用化合物和蛋白质序列以及分子图的模型相比并没有显著性能提升。
- 对于化合物-蛋白质结合对的表征,非共价相互作用的表示很重要。结合位点有助于理解非共价相互作用。目前DeepSite,DeepSurf已经将深度学习用于结合位点预测。
- 利用注意力的解释不够全面,在大规模数据集中会出现不一致的解释结果。
实际应用
CPI预测是寻找苗头化合物的第一步。为了加快苗头化合物的发现过程,DeepScreening根据结合亲和力针对特定靶标进行大规模化合物筛选。深度学习可以加快发现新的活性化合物,推进药物发现。