🌞欢迎来到图神经网络的世界
🌈博客主页:卿云阁💌欢迎关注🎉点赞👍收藏⭐️留言📝
🌟本文由卿云阁原创!
📆首发时间:🌹2024年3月20日🌹
✉️希望可以和大家一起完成进阶之路!
🙏作者水平很有限,如果发现错误,请留言轰炸哦!万分感谢!
目录
GNN起源
图的矩阵表示
层内与层间的消息传递
GCN
GraphSAGE
代码实战
GAT
代码实战
GNN起源
(1)数学中的空间有很多种,大部分都是定义在欧氏里德空间的,比如图像,文本。除此之外还存在着大量的非欧空间,比如分子结构。
(2) 图嵌入常⻅模型有DeepWalk,Node2Vec等,然而,这些方法方法有两种严重的缺点,首先就是节点编码中权重未共享,导致权重数量随着节点增多而线性增大,另外就是直接嵌入方法缺乏泛化能力,意味着无法处理动态图以及泛化到新的图。
如何把这种图结构嫁接到神经网络上,图神经网络就诞生了。和传统的神经网络结构相比,它解决了两个问题。
- 图结构的矩阵画表示
- 层内与层间的消息传递
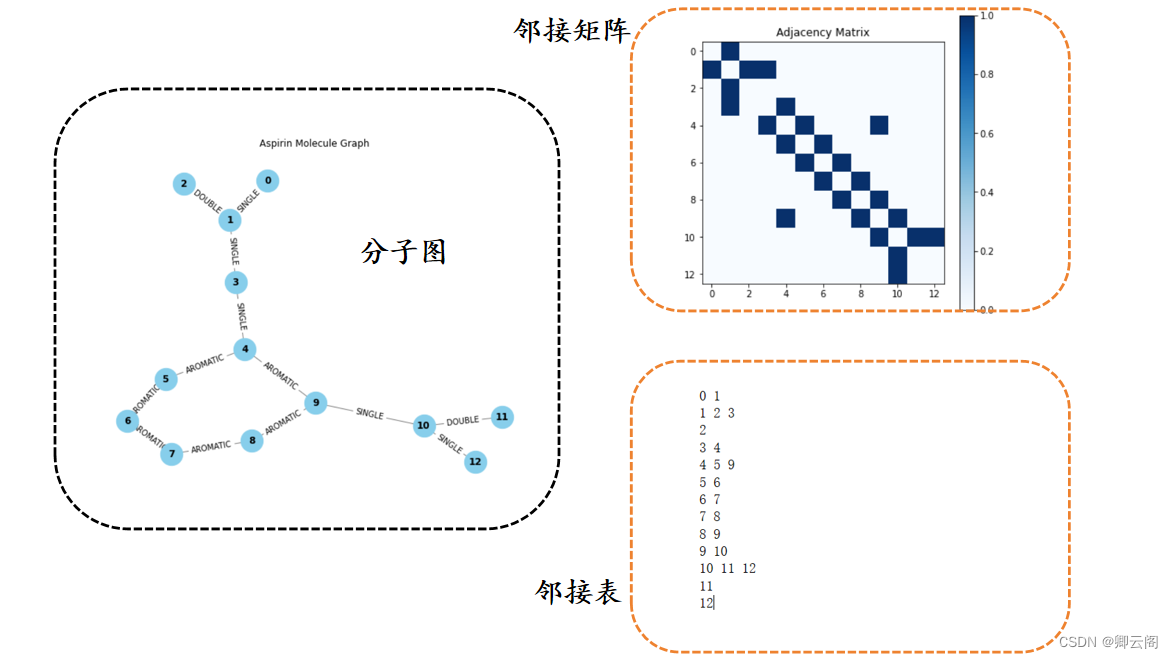
图的矩阵表示
- 借用邻接矩阵
- 考虑稀疏性,还可以使用邻接表。
层内与层间的消息传递
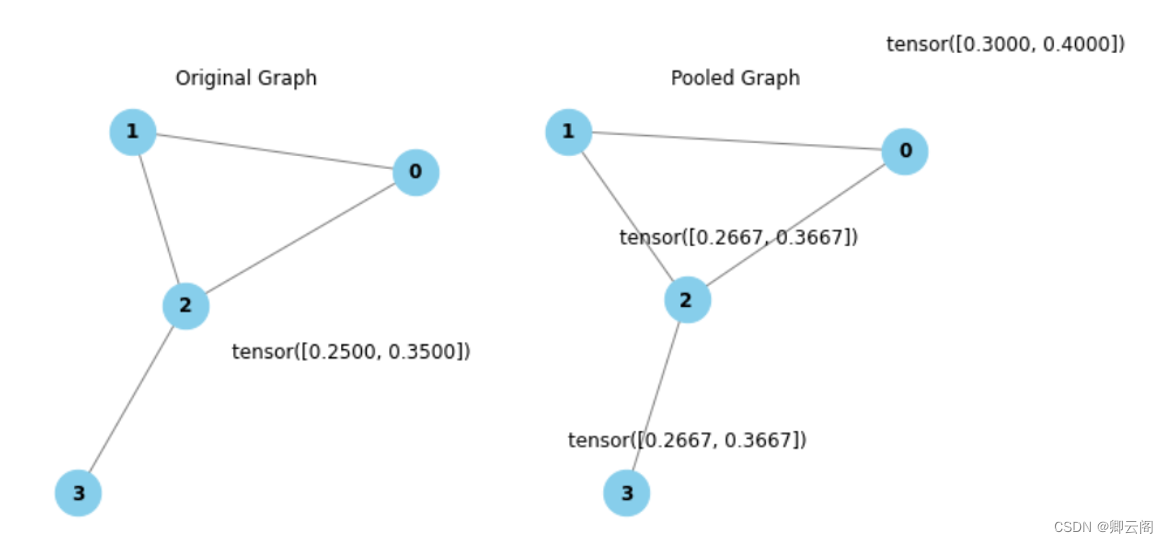
聚合
简单来说一个节点或者边的特征,不光看它自己,还要由它相邻元素的加权求和决定。层内的聚合常常被称之为池化。
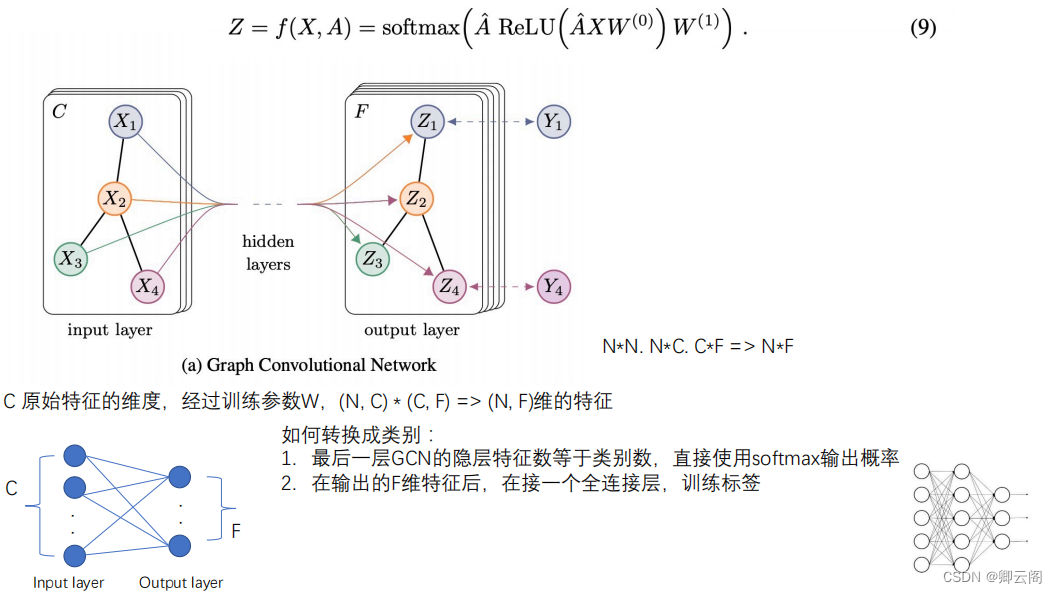
层级间的关系传递,通过节点的连接关系进行,也可以看成是一种聚合,根据聚合方法的差异形成了不同的算法,最简单的是图卷积网络GCN。就是在层间经过邻域聚合实现卷积特征提取。左乘于邻接矩阵表示对每个节点来说,该节点的特征为邻域节点的特征,相加之后的结果。
如果聚合的时候没有用全部的邻域节点,而是先采样再聚合,就是GraphSAGE算法。
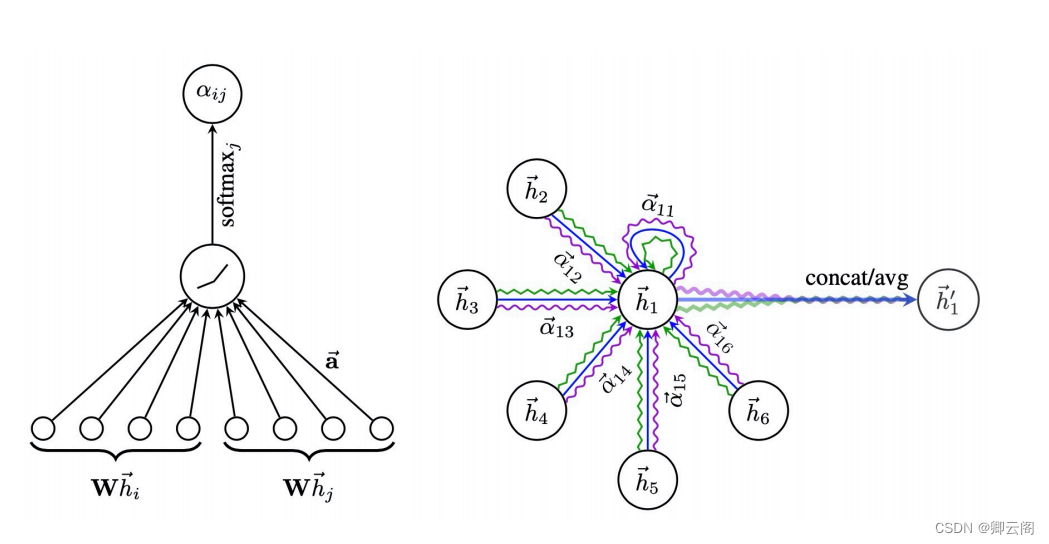
如果聚合的时候考虑了领域节点的权重,也就是运用了注意力机制,那么就是图注意力网络GAT。
聚合还可以用在非监督模型上,比如把图和变自分编码器相结合,形成GAE算法。
除此之外还有更复杂的图生成网络,和图时空网络。
GCN
原理解析:
代码实战:
import torch import torch.nn as nn import dgl import dgl.function as fn import networkx as nx import matplotlib.pyplot as plt from rdkit import Chem from rdkit.Chem import Draw # 构建阿司匹林分子 aspirin_smiles = "CC(=O)OC1=CC=CC=C1C(=O)O" aspirin_mol = Chem.MolFromSmiles(aspirin_smiles) # 构建分子图 aspirin_graph = dgl.from_networkx(nx.Graph(Chem.rdmolops.GetAdjacencyMatrix(aspirin_mol))) # 可视化分子结构 Draw.MolToImage(aspirin_mol) # 定义GCN模型 class GCN(nn.Module): def __init__(self, in_feats, hidden_size, num_classes): super(GCN, self).__init__() self.conv1 = dgl.nn.GraphConv(in_feats, hidden_size) self.conv2 = dgl.nn.GraphConv(hidden_size, num_classes) def forward(self, g, features): h = self.conv1(g, features) h = torch.relu(h) h = self.conv2(g, h) return h # 初始化GCN模型 input_dim = 1 # 输入特征维度为1,因为我们只考虑一个原子的属性 hidden_size = 64 num_classes = 2 # 为简单起见,假设我们的任务是二分类 gcn_model = GCN(input_dim, hidden_size, num_classes) # 可视化GCN模型结构 print(gcn_model) # 可视化分子图 plt.figure(figsize=(8, 6)) nx.draw(aspirin_graph.to_networkx(), with_labels=True, node_color='skyblue', node_size=800, font_size=12, font_weight='bold', edge_color='gray') plt.title('Molecular Graph') plt.show()
GraphSAGE
代码实战
我们来实现了一个简单的 GraphSAGE 模型,并对阿司匹林的分子结构进行预测。首先,我们需要构建一个简单的图结构来表示阿司匹林的分子。然后,我们将定义一个GraphSAGE 模型,并使用该模型对阿司匹林分子的属性进行预测。
import torch import torch.nn as nn import dgl import dgl.function as fn import networkx as nx import matplotlib.pyplot as plt import numpy as np # 构建一个简单的分子图来表示阿司匹林的结构 aspirin_graph = dgl.graph(([0, 1, 1, 2], [1, 0, 2, 1])) # 定义边的连接关系 # 可视化分子图 plt.figure(figsize=(4, 4)) nx.draw(aspirin_graph.to_networkx(), with_labels=True, node_color='skyblue', node_size=800, font_size=12, font_weight='bold', edge_color='gray') plt.title('Molecular Graph') plt.show() # 定义GraphSAGE模型 class GraphSAGE(nn.Module): def __init__(self, in_feats, hidden_size, num_classes): super(GraphSAGE, self).__init__() self.conv1 = dgl.nn.SAGEConv(in_feats, hidden_size, 'mean') self.conv2 = dgl.nn.SAGEConv(hidden_size, num_classes, 'mean') def forward(self, g, features): h = self.conv1(g, features) h = torch.relu(h) h = self.conv2(g, h) return h # 初始化GraphSAGE模型 input_dim = 1 # 输入特征维度为1,因为我们只考虑一个原子的属性 hidden_size = 64 num_classes = 2 # 为简单起见,假设我们的任务是二分类 graphsage_model = GraphSAGE(input_dim, hidden_size, num_classes) # 生成随机的示例数据 num_samples = aspirin_graph.number_of_nodes() node_features = torch.randn(num_samples, input_dim) # 随机生成二分类标签(示例) labels = torch.randint(0, 2, (num_samples,)) # 将标签添加到图中的节点 aspirin_graph.ndata['features'] = node_features aspirin_graph.ndata['labels'] = labels # 定义损失函数 loss_fn = nn.CrossEntropyLoss() # 模型训练 optimizer = torch.optim.Adam(graphsage_model.parameters(), lr=0.001) epochs = 50 for epoch in range(epochs): logits = graphsage_model(aspirin_graph, aspirin_graph.ndata['features']) loss = loss_fn(logits, aspirin_graph.ndata['labels']) optimizer.zero_grad() loss.backward() optimizer.step() if (epoch + 1) % 10 == 0: print(f'Epoch [{epoch+1}/{epochs}], Loss: {loss.item()}') # 使用模型进行预测(示例) with torch.no_grad(): predicted_labels = torch.argmax(graphsage_model(aspirin_graph, aspirin_graph.ndata['features']), dim=1) print("Predicted Labels:", predicted_labels)
GAT
代码实战
import torch import torch.nn as nn import dgl import dgl.function as fn import networkx as nx import matplotlib.pyplot as plt # 构建阿司匹林分子的简单图结构 aspirin_graph = dgl.graph(([0, 0, 0, 1, 2], [1, 2, 3, 3, 3])) # 使用边列表构建图 # 定义节点特征 node_features = torch.tensor([ [0.1, 0.2], [0.2, 0.3], [0.3, 0.4], [0.4, 0.5] ], dtype=torch.float) # 将节点特征设置到图中 aspirin_graph.ndata['feat'] = node_features # 可视化分子图 plt.figure(figsize=(8, 6)) nx.draw(aspirin_graph.to_networkx(), with_labels=True, node_color='skyblue', node_size=800, font_size=12, font_weight='bold', edge_color='gray') plt.title('Molecular Graph') plt.show()class GAT(nn.Module): def __init__(self, in_dim, hidden_dim, out_dim, num_heads): super(GAT, self).__init__() self.conv1 = dgl.nn.GATConv(in_dim, hidden_dim, num_heads) self.conv2 = dgl.nn.GATConv(hidden_dim * num_heads, out_dim, num_heads) def forward(self, g, features): h = self.conv1(g, features) h = torch.relu(h) h = self.conv2(g, h) return h # 初始化 GAT 模型 input_dim = 2 # 输入特征维度 hidden_dim = 64 out_dim = 1 # 输出维度,这里假设我们只需要一个输出维度进行二分类 num_heads = 2 gat_model = GAT(input_dim, hidden_dim, out_dim, num_heads) # 输出 GAT 模型结构 print(gat_model)
【Deep Learning 11】Graph Neural Network
news2026/3/31 20:01:33











本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。如若转载,请注明出处:http://www.coloradmin.cn/o/1556665.html
如若内容造成侵权/违法违规/事实不符,请联系多彩编程网进行投诉反馈,一经查实,立即删除!相关文章
openLooKeng开发环境搭建
文章目录 搭建OpenLooKeng开发环境要求 以下是搭建OpenLooKeng开发环境的基本步骤:1、从OpenLooKeng的GitHub仓库克隆代码:2、 构建OpenLooKeng生成IntelliJ IDEA项目文件 airbase构建项目过程中出现的问题checkstyle错误版本冲突问题hetu-heuristic-ind…

辽宁政府采购网怎么入驻?
辽宁政府采购网的入驻流程包括以下几个主要步骤:
注册账号:在辽宁政府采购网上商城注册账号。CA证书领取:注册成功后,需要领取CA证书以登录网上商城。搭建自营商城:因为后期需要和辽宁政府采购网上商城进行入驻&#…
执行 kubeadm join 报错ERROR FileAvailable--etc-kubernetes-kubelet.conf
执行 kubeadm join 报错ERROR FileAvailable–etc-kubernetes-kubelet.conf
[rootk8snode2 ~]# kubeadm join apiserver.demo:6443 --token c4nezq.ecv2kg9ok6gsresw --discovery-token-ca-cert-hash sha256:be1a55bea6b5bb5c8810434d3905a9cd0bbc33181862f7ad601346e1ab0…
.NET CORE 分布式事务(二) DTM实现TCC
目录
引言:
1. TCC事务模式
2. TCC组成
3. TCC执行流程
3.1 TCC正常执行流程
3.2 TCC失败回滚
4. Confirm/Cancel操作异常
5. TCC 设计原则
5.1 TCC如何做到更好的一致性
5.2 为什么只适合短事务
6. 嵌套的TCC
7. .NET CORE结合DTM实现TCC分布式事务 …
wireshark创建显示过滤器实验简述
伯克利包过滤是一种在计算机网络中进行数据包过滤的技术,通过在内核中插入过滤器程序来实现对网络流量的控制和分析。 在数据包细节面板中创建显示过滤器,显示过滤器可以在wireshark捕获数据之后使用。
实验拓扑图: 实验基础配置࿱…

计算机专业在找工作时的注意事项
目录 说在前面关于我一些忠告关于简历关于银行写在最后 说在前面
满满的求生欲。我不是什么大佬,更没有能力教大家什么。只是看到有不少学弟学妹,还在为找一份工作焦头烂额,却没有努力的方向。所以这里斗胆给计算机相关专业的学弟学妹们的一…

【动手学深度学习-pytorch】 9.4 双向循环神经网络
在序列学习中,我们以往假设的目标是: 在给定观测的情况下 (例如,在时间序列的上下文中或在语言模型的上下文中), 对下一个输出进行建模。 虽然这是一个典型情景,但不是唯一的。 还可能发生什么其…
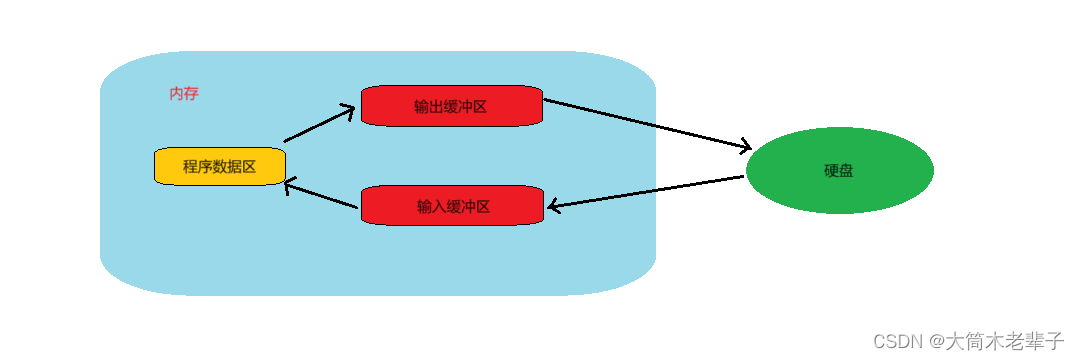
文件操作(随机读写篇)
1. 铺垫
建议先看:
文件操作(基础知识篇)-CSDN博客
文件操作(顺序读写篇)-CSDN博客
首先要指出的是,本篇文章中的“文件指针”并不是指FILE*类型的指针,而是类似于打字时的光标的东西。
打…
EMD关于信号的重建,心率提取
关于EMD的俩个假设: IMF 有两个假设条件: 在整个数据段内,极值点的个数和过零点的个数必须相等或相差最多不能超过一 个;在任意时刻,由局部极大值点形成的上包络线和由局部极小值点形成的下包络线 的平均值为零&#x…

Ubuntu上安装d4rl数据集
Ubuntu上安装d4rl数据集
D4RL的官方 github: https://github.com/Farama-Foundation/D4RL
一、安装Mujoco
1.1 官网下载mujoco210文件
如果装过可以跳过这步 链接:https://github.com/deepmind/mujoco/releases/tag/2.1.0
下载第一个文件即可。我这里是在windo…

【JAVA】精密逻辑控制过程(分支和循环语句)
✅作者简介:大家好,我是橘橙黄又青,一个想要与大家共同进步的男人😉😉 🍎个人主页: 橘橙黄又青-CSDN博客 目标: 1. Java 中程序的逻辑控制语句 2. Java 中的输入输出方式 3. 完成…
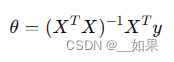
动手学机器学习线性回归+习题
线性回归
矩阵求导: 左边是分子布局,右边是分母布局,一般都用分母布局 解析解与数值解:
解析解是严格按照公式逻辑推导得到的,具有基本的函数形式。给出任意的自变量就可以求出其因变量
数值解是采用某种计算方法&a…

写作类AI推荐(二)
本章要介绍的写作AI如下: 火山写作 主要功能: AI智能创作:告诉 AI 你想写什么,立即生成你理想中的文章AI智能改写:选中段落句子,可提升表达、修改语气、扩写、总结、缩写等文章内容优化:根据全文…

【C++】string类(常用接口)
🌈个人主页:秦jh__https://blog.csdn.net/qinjh_?spm1010.2135.3001.5343🔥 系列专栏:http://t.csdnimg.cn/eCa5z 目录
修改操作 push_back
append operator
assign
insert
erase
replace
c_str
find
string类非成…
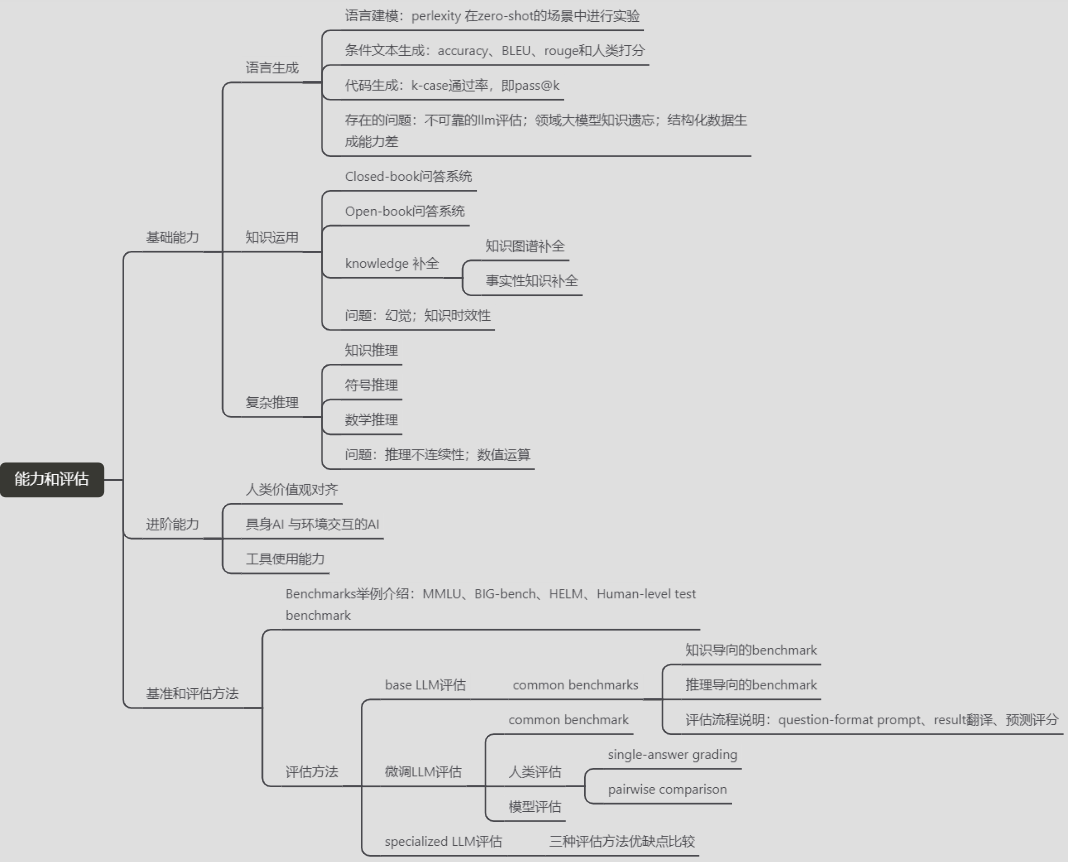
【ReadPapers】A Survey of Large Language Models
LLM-Survey的llm能力和评估部分内容学习笔记——思维导图 思维导图
参考资料
A Survey of Large Language Models论文的github仓库
【AcWing】蓝桥杯集训每日一题Day8|日期问题|前缀和|3498.日期差值(C++)
3498.日期差值
3498. 日期差值 - AcWing题库难度:简单时/空限制:1s / 64MB总通过数:5763总尝试数:18345来源:上海交通大学考研机试题算法标签模拟日期问题 题目内容
有两个日期,求两个日期之间的天数&…
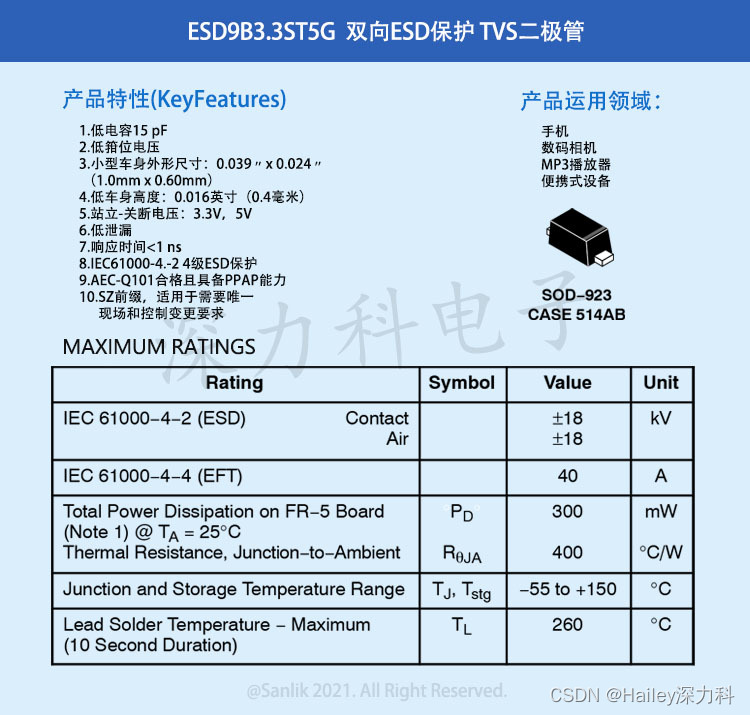
ESD保护二极管ESD9B3.3ST5G 以更小的空间实现强大的保护 车规级TVS二极管更给力
什么是汽车级TVS二极管?
TVS二极管是一种用于保护电子电路的电子元件。它主要用于电路中的过电压保护,防止电压过高而损坏其他部件。TVS二极管通常被称为“汽车级”是因为它们能够满足汽车电子系统的特殊要求。
在汽车电子系统中,由于车辆启…
