目录
基础知识
实操
层次聚类
划分聚类
方法一:K均值聚类(最常见)
方法二:基于中心点的划分(PAM)
避免不存在的类
基础知识
概念:
聚类分析是一种数据归约技术,旨在揭露一个数据集中观测值的子集。例如:对DNA微阵列数据进行聚类分析获得基因表达模型,从而帮助更好的理解疾病的原因;基于抑郁症病人的症状和人口学统计数据对病人进行聚类,试图得出抑郁症的亚型。
聚类的分类:
1.层次聚类:每一个观测值自成一类,这些类每次两两合并,知道所有类被聚成一类。
常用的算法: 单联动(single linkage)、全联动(complete linkage)、平均联动(average linkage)、质心(centroid)和Ward 方法。
2.划分聚类:首先指定类的个数K,然后观测值随机分成K类,再重新形成聚合的类。
常用的算法:K均值(K-means)和围绕中心的划分(PAM)
聚类的步骤:
(1)选择合适的变量
选择你感觉可能对识别和理解数据中不同观测值分组有重要影响的变量
(2)缩放数据
分析中如果选择的变量变化范围很大,那么该变量对结果的影响也是最大的,这是不可取的。最常用的缩放数据的方法是将每个变量的标准化为均值为0标准差为1的变量。其他方法:每个变量被其最大值相除或该变量减去它的平均值并除以变量的平均绝对偏差。
三种方法代码:
df1 <- apply(mydata,2,function(x)((x-mean(x))/sd(x)))
df2 <- apply(mydata,2,function(x)(x/max(x)))
df3 <- apply(mydata,2,function(x)((x-mean(x))/mad(x)))(3)寻找异常点
聚类分析对异常值非常敏感,可以通过outlier包中的函数筛选和删除异常单变量离群点。mvoutlier包中包含了能识别多元变量的离群点的函数。围绕中心的划分是一种对异常值稳健的聚类方法。
(4)计算距离
需要计算被聚类的实体之间的距离。两个观测值之间最常用的距离量度是欧几里得距离,其他可以选择的量度包括曼哈顿距离、兰氏距离、非对称二元距离、最大距离和闵可夫斯基距离。
(5)选择聚类算法
层次聚类对于小样本来说很实用(如150个观测值或更少),划分的方法能处理更大的数据量,但需要先确定聚类的个数。
(6)获得一种或多种聚类方法
(7)确定类的数目
常用的方法:尝试不同的类数并比较解的质量,NbClust包中NbClust()函数提供30个不同的指标来帮助进行选择。
(8)获得最终的聚类解决方案
(9)结果可视化
层次聚类的结果通常表示为一个树状图,划分聚类的结果通常利用可视化双变量聚类图来表示。
(10)解读类
(11)验证结果
实操
层次聚类
案例:flexclust包中营养数据nutrient作为层次聚类,以期望回答以下问题:
- 基于五种营养标准的27种鱼,禽、肉的相同点和不同点是什么?
- 是否有一种方法将这些食物分成若干个有意义的类?
载入数据:
#install.packages("flexclust")
data(nutrient,package = "flexclust")
head(nutrient)
计算距离:
欧几里得距离:
在图上是用点的 N 个属性构成两个 N 维向量并计算欧几里得距离。
其中,xi1 表示第一个点的第 i 维坐标,xi2 表示第二个点的第 i 维坐标。
欧几里得距离的取值范围是 [0,+∞],数值越小越相似。

dist()函数能用来计算矩阵或数据框的中所有行之间的距离,格式为dist(x,method=),x表示输入数据,默认欧几里得距离。
d <- dist(nutrient)
as.matrix(d)[1:4,1:4]
观测值之间的距离越大,说明异质性越大。观测值和它自己之间的距离为0。
注意:
欧几里得距离通常作为连续型数据的距离度量。但是如果存在其他类型的数据,则需要相异的替代措施。使用cluster包中的display()函数来获得包含任意二元(binary)、名义(nominal)、有序(ordinal)、连续(continunous)属性组合的相异矩阵。cluster包中agnes()函数可以进行层次聚类,pam()函数可以进行围绕中心点的划分的方法。
缩放数据:
从数据中可以看到energy这个变量变化范围比较大,对距离的影响大。所以缩放数据有利于平衡各变量的影响。
nutrient1 <- apply(nutrient,2,function(x)((x-mean(x))/sd(x)))
d1 <- dist(nutrient1)
as.matrix(d1)[1:4,1:4]将每个变量标准化为均值为0,标准差为1的变量。

层次聚类:
该案例为27个样本,5个变量,属于小样本数据。因此使用层次聚类
思想:每一个实例或观测值属于一类,聚类就是每一次把两类聚成新的一类,直到所有的类聚完成为一个单个类为止。
算法步骤:
- 定义每个观测值(行或单元)为一类;
- 计算每类和其他类的距离;
- 把距离最短的两类合并为一类,这样类的个数就减少一个;
- 重复步骤2和3,直到包含所有的观测值的类合并成为单个的类为止。
| 聚类方法 | 两类之间的距离定义 |
| 单联动 | 一个类中的点和另一个类中的点的最小距离 |
| 全联动 | 一类中的点和另一个类中的点的最大距离 |
| 平均联动 | 一类中的电荷另一类中的点的平均距离(UPGMA,非加权对组平均) |
| 质心 | 两类中质心(变量均值向量)之间的距离。对于单个的观测值来说,质心就是变量的值 |
| Ward法 | 两个类之间所有变量的方差分析的平方和 |
单联动倾向于发现细长的、雪茄型的类;全联动聚类倾向于发现大致相等的直径紧凑类,对异常值敏感。平均联动折中,倾向于把方差小的类聚合。ward法倾向于将少数观测值的类聚合在一起,倾向于产生跟观测值个数大致相等的类,对异常值敏感。质心法对异常值不是很敏感,但不如ward法和平均联动表现好。
层次聚类使用函数hclust()实现,hclust(d,method=),d是dist()函数产生的距离矩阵,method包括以上五种,"single", "complete", "average", "centroid", "ward"
fit.average <- hclust(d1,method = "average")
plot(fit.average,hang=-1,cex=0.8,main = "Average Linkage Clustering")hang的意义是展示观测值标签,让他们挂在0的下面
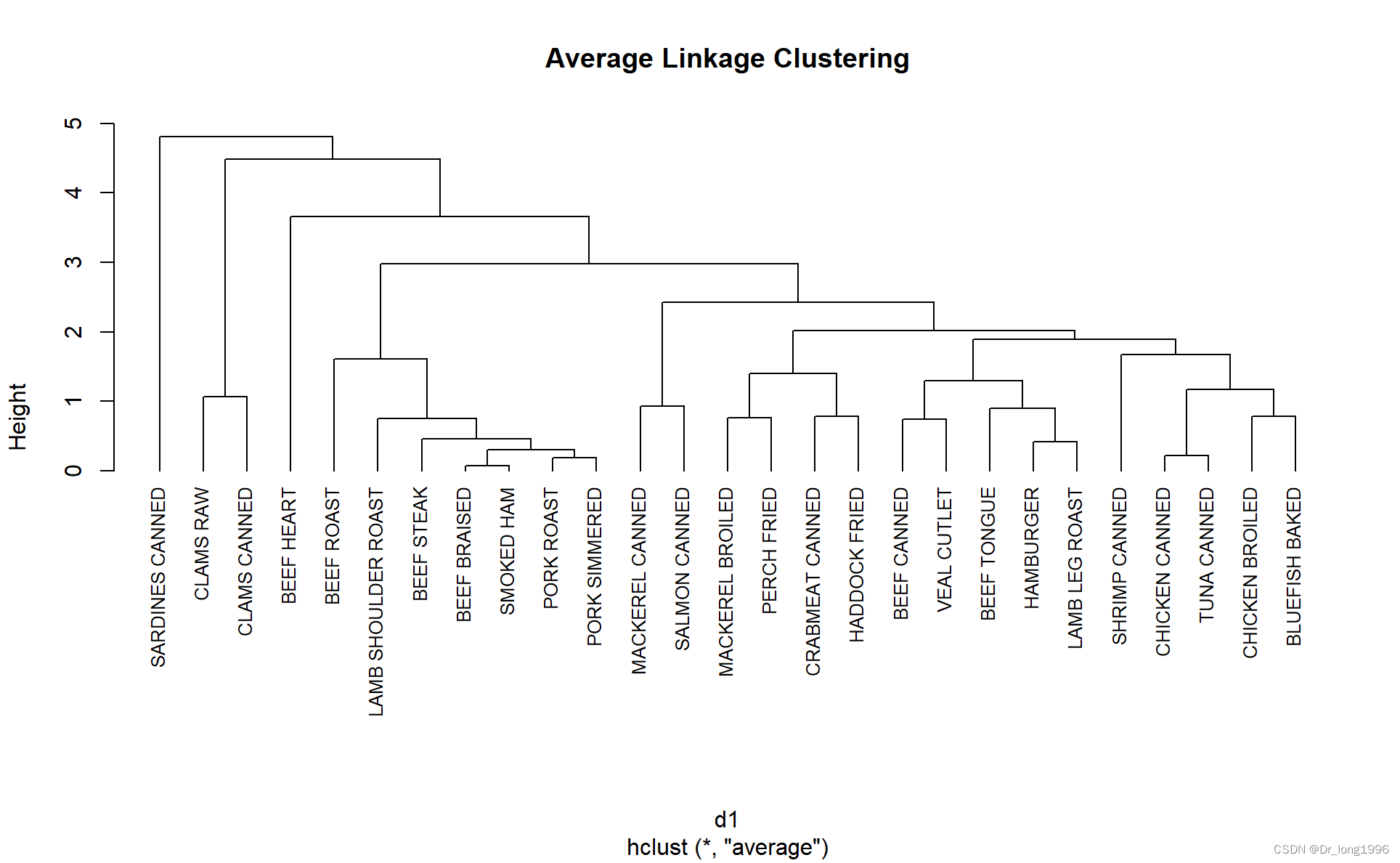
高度表示该高度之间合并的判定值。对于平均联动来说,标准就是一类中点和其他类中点的距离平均值。
结果解读:
可以从图中看到食物之间的相似性/异质性,比如tuna canned和chicken canned是相似的,但都和clams canned有很大的不同。
确定类的数目:
NbClust包中提供了众多的指数来确定一个聚类分析中类的最佳数目。
library(NbClust)
nc <- NbClust(nutrient1,distance = "euclidean",
min.nc = 2,max.nc = 15,
method = "average")需要做聚类的矩阵或数据框,distance指定距离测度,method指定聚类方法,min.nc和max.nc最小最大的聚类个数。他返回每一个聚类指数,建议聚类的最佳数目。
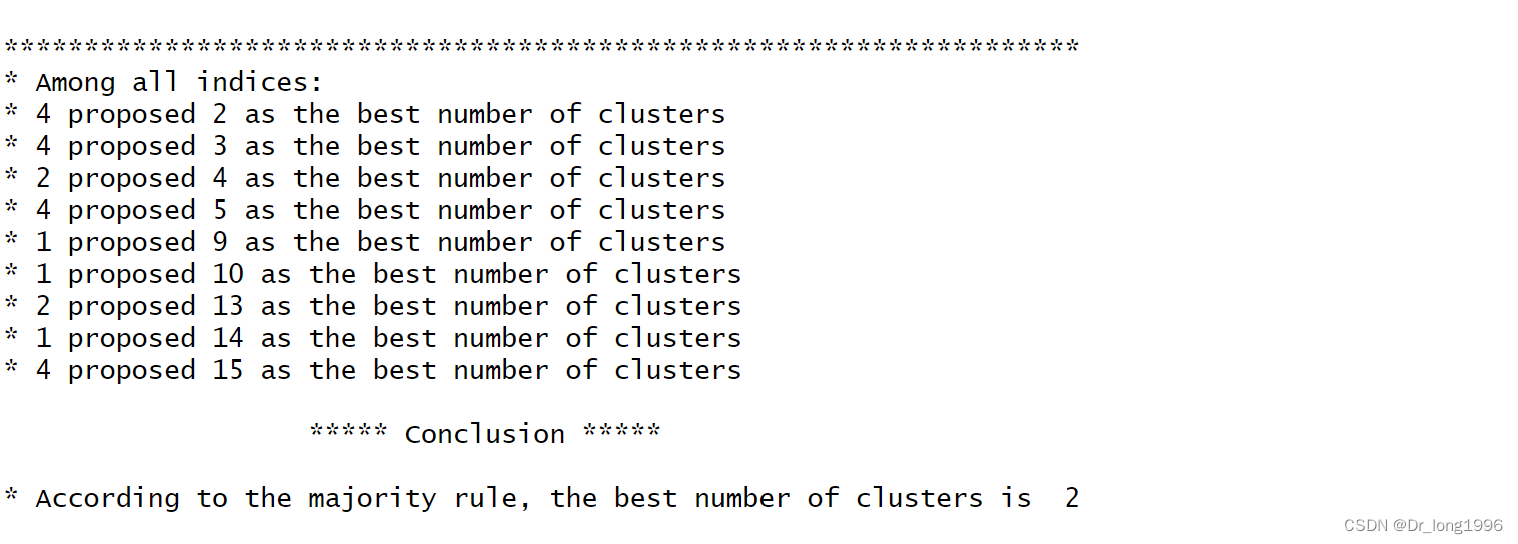
四个人赞同2,四个判定准则赞同聚类个数为3,4个赞同5,4个赞同15,选择其中一个解释其中意义。
#将聚类结果分成5类
clusters <- cutree(fit.average,k=5)
#查看每一类的数目。
table(cluster)
#原始数据每一类的变量特征
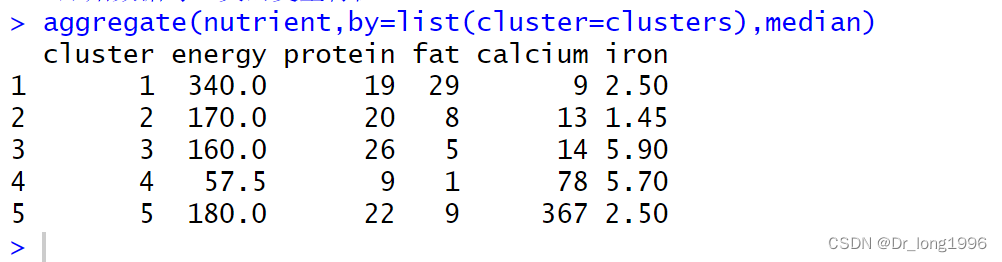
aggregate(nutrient,by=list(cluster=clusters),median)
#缩放数据每一类的变量特征
aggregate(nutrient1,by=list(cluster=clusters),median)获取每一类的中位数

绘制聚类图:
plot(fit.average,hang=-1,cex=.8,
main = "Average Linkage Clustering\n5 Cluster Solution")
rect.hclust(fit.average,k=5)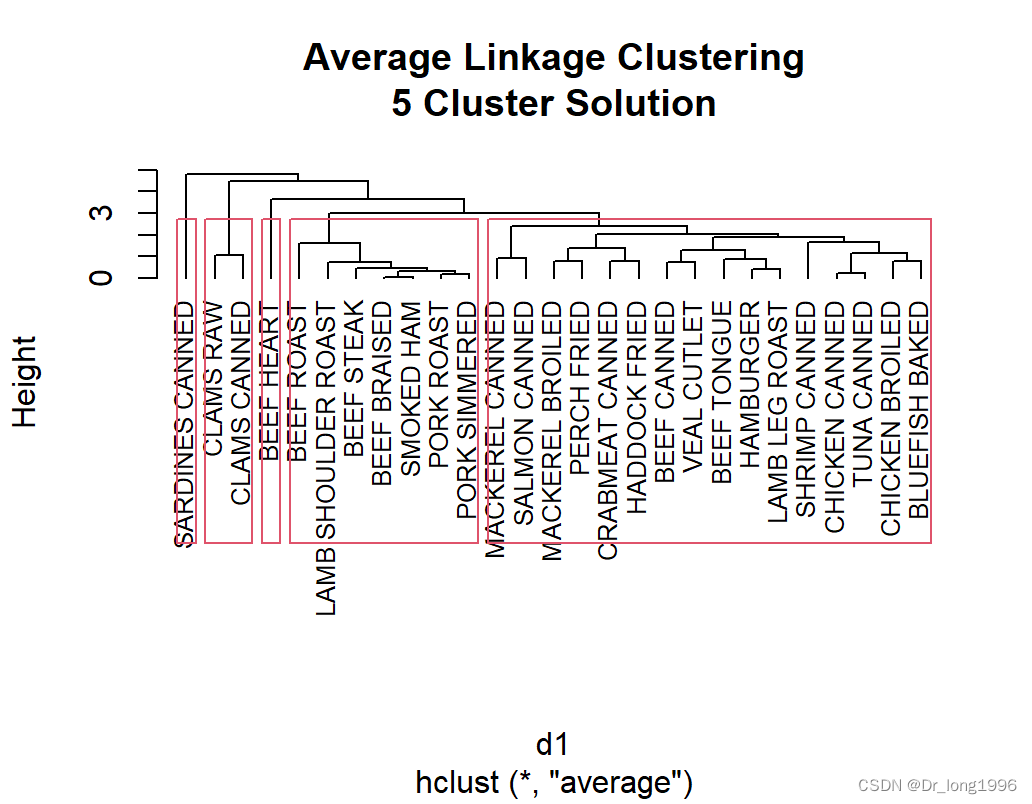
sardines canned自己形成一类,因为钙含量比其他组高了很多。beef heart也单独成为一类,因为富含蛋白质和铁。clams类的是低蛋白和高铁。
划分聚类
划分方法中,观测值被分为K组并根据给定的规则改组成为最有粘性的类。
案例:178种意大利葡萄酒样品的13种化学成分。这些意大利葡萄酒中样品能继续分成为更细的组吗?如果能,分多少组?他们的特征是什么?
方法一:K均值聚类(最常见)
思想:
- 随机选择K行,即K个中心点;
- 把每个数据点分配到离它最近的中心点;
- 重新计算每类中的点到该类中心点距离的平均值;
- 分配每个数据到它最近的中心点;
- 重复步骤3和4直到所有的观测点不再被分配,或是达到最大的迭代次数(R把10作为默认迭代次数)
优势:K均值聚类能处理比层次聚类更大的数据集。观测值不会永远被分到一类中。
格式:Kmeans(X,centers),X表示数值数据集(矩阵或数据框),centers是要提取的聚类数目。函数返回类的成员,类中心,平方和,和类大小
聚类方法对初始中心值的选择也非常敏感,nstart选项尝试多种初始配置并输出最好的一个。例如nstart=25会生成25个初始配置。
#install.packages("rattle")
data(wine,package = "rattle")
head(wine)
在数据集中,观测值代表三种葡萄酒的品种,Type则是表示三种不同类型。因为聚类,看看是否能识别出三种不同的类型。
缩放数据:
df <- scale(wine[-1])变量值变化比较大,所以在聚类前进行标准化。
确定聚类的个数:
library(NbClust)
set.seed(1234)
devAskNewPage(ask=T)
nc <- NbClust(df,min.nc = 2,max.nc = 15,method = "kmeans")
table(nc$Best.nc[1,])
barplot(table(nc$Best.nc[1,]),
xlab = "Number of Clusters",ylab = "Number of Criteria",
main = "Number of Clusters Chosen by 26 Criteria")

在NbCluster包中26个指标中有19个指标建议使用类别为3的聚类方案。,并非30个指标都可以计算每个数据集。
进行K均值聚类分析
set.seed(1234)
fit.km <- kmeans(df,3,nstart = 25)
fit.km$size
fit.km$centers
第一个是每一个分类中的样本数,第二个是每一类中每个指标的标准化均值,如果计算原始均值,可以使用aggregate()函数
aggregate(wine[-1],by=list(cluster=fit.km$cluster),mean)
评价
K均值很好的反映了类型变量中真正的数据结构吗?交叉列联表和类别进行比较
#为了区别分类结果和实际的类型,重新赋值
wine$Type <- ifelse(wine$Type==1,"A",
ifelse(wine$Type==2,"B","C"))
table(wine$Type,fit.km$cluster)
量化类型变量与类之间的协议:flexclust包中兰德指数Rank index
library(flexclust)
randIndex(table(wine$Type,fit.km$cluster))
调整的兰德指数为两种划分提供了一种衡量两个分区之间的协定,即调整后机会的量度。它的变化范围是从-1(不同意)到1(完全同意)。
方法二:基于中心点的划分(PAM)
因为K-means聚类方法是基于均值的,所以它对异常值是敏感的。一个更稳健的方法是围绕中心点的划分(PAM)。K-means聚类一般使用欧几里得距离,而PAM可以使用任意的距离来计算。因此,PAM可以容纳混合数据类型,并且不仅限于连续变量。
PAM算法如下:
- 随机选择K个观测值(每个都称为中心点);
- 计算观测值到各个中心的距离/相异性;
- 把每个观测值分配到最近的中心点;
- 计算每个中心点到每个观测值的距离的总和(总成本);
- 选择一个该类中不是中心的点,并和中心点互换;
- 重新把每个点分配到距它最近的中心点;
- 再次计算总成本;
- 如果总成本比步骤(4)计算的总成本少,把新的点作为中心点;
- 重复步骤(5)~(8)直到中心点不再改变。
可以使用cluster包中的pam()函数使用基于中心点的划分方法。格式如下:
pam(x, k, metric="euclidean", stand=FALSE)
x表示数据矩阵或数据框,k表示聚类的个数,metric表示使用的相似性/相异性的度量,而stand是一个逻辑值,表示是否有变量应该在计算该指标之前被标准化。
library(cluster)
set.seed(1234)
fit.pam <- pam(wine[-1],k=3,stand = T)
fit.pam$medoids
这里得到的中心点是葡萄酒数据集中实际的观测值。

这里分别选择了36,107,175观测值来代表三类。

ct.pam <- table(wine$Type,fit.pam$clustering)
randIndex(ct.pam) 但是,PAM在本例中的表现不如K均值,函数randIndex()结果下降为了0.699。
但是,PAM在本例中的表现不如K均值,函数randIndex()结果下降为了0.699。
避免不存在的类
聚类分析是一种旨在识别数据集子组的方法,并且 在此方面十分擅长。事实上,它甚至能发现不存在的类。 代码中提供一个对完全随机数据进行PAM聚类的例子。图展示了这个数据集的分布情况。其中并不存在聚类。
#install.packages("fMultivar")
library(fMultivar)
set.seed(1234)
df <- rnorm2d(1000,rho = 0.5)
df <- as.data.frame(df)
plot(df,main="Bivariate Normal Distribution with rho=0.5")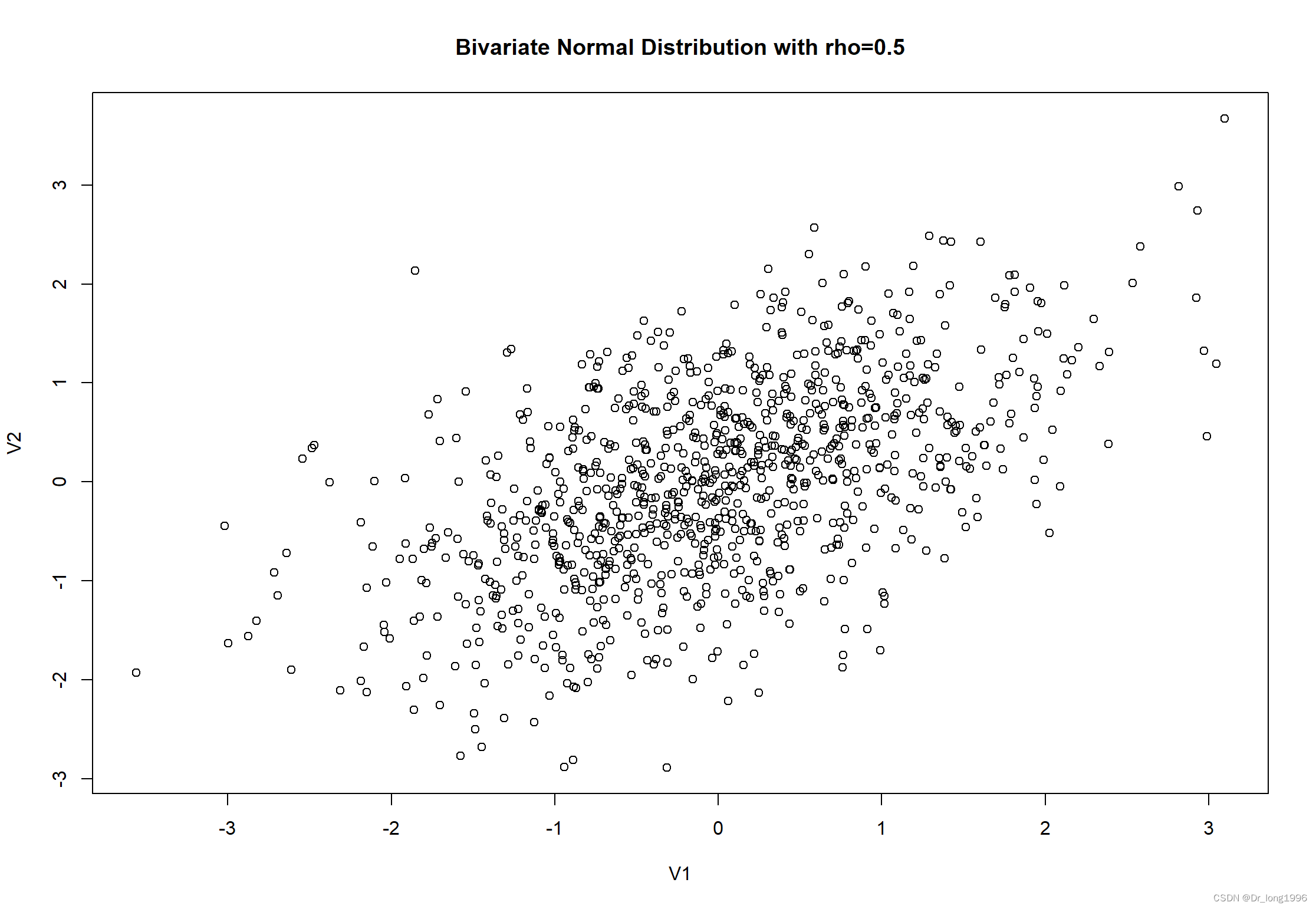
library(NbClust)
nc <- NbClust(df, min.nc=2, max.nc=15, method="kmeans")
dev.new()
barplot(table(nc$Best.n[1,]),
xlab="Numer of Clusters", ylab="Number of Criteria",
main ="Number of Clusters Chosen by 26 Criteria")
NbClust的结果都建议分类为2或3。利用PAM方法进行双聚类结果
library(ggplot2)
library(cluster)
fit <- pam(df,k=2)
df$clustering <- factor(fit$clustering)
ggplot(data=df,
aes(x=V1,y=V2,
color=clustering,shape=clustering))+
geom_point()+
ggtitle("Clustering of Bivariate Normal Data")
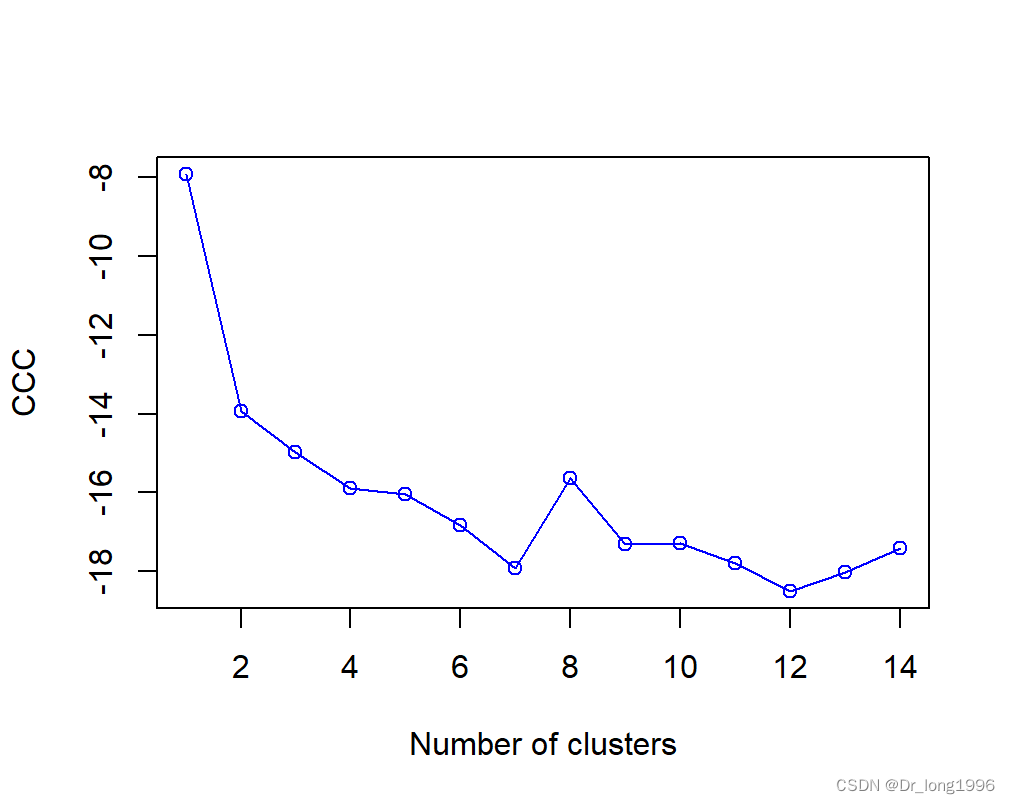
很明显划分是人为的。实际上在这里没有真实的类。NbClust包中的立方聚类规则(Cubic Cluster Criteria,CCC)往往可以帮助我们揭示不存在的结构。当CCC的值为负并且对于两类或是更多的类递减时,就是典型的单峰分布(如图)。
plot(nc$All.index[,4],type="o",ylab="CCC",
xlab="Number of clusters",col="blue")