今天为大家介绍的是来自Tehmina Masud, Amit Deshwar, Shreshth Gandhi, Brendan J. Frey团队的一篇论文。精确地对RNA生物学进行建模和预测一直是一个长期存在的挑战,对于变异解释和定制治疗的制定具有重要的临床意义。作者提出了一个RNA生物学的基础模型,名为“BigRNA”,它经过了数千个基因组匹配数据集的训练,可以从DNA序列预测组织特异性的RNA表达、剪接、microRNA位点以及RNA结合蛋白的特异性。

建立能够从DNA序列预测基因表达的机器学习模型一直是长期以来的科研界的研究目标,最近深度学习的进展使这个目标取得了显著的进展。这些模型可以通过确定致病基因变异如何改变基因表达和基因加工,以及设计定制的药物候选物来彻底改革药物发现过程。目前,大多数研究工作都集中在预测衡量整体基因表达水平的数据上,这些数据并不适合预测调控干预。RNA测序(RNA-seq)数据提供了一个广泛可用的资源,用于高分辨率测量RNA表达并捕捉不同基因型之间的复杂转录调控事件。这包括在RNA-seq数据本身中固有编码的外显子变异,以及通过资源丰富的项目,如Genotype-Tissue Expression(GTEx)项目,将RNA-seq与全基因组测序(WGS)相结合。虽然建立直接从RNA-seq学习的深度神经网络为我们提供了理解DNA序列变化如何导致复杂转录表型变化的机会,但这个目标一直难以实现。
作者提出入了一个名为“BigRNA”的深度学习模型,它直接在RNA-seq数据集上进行训练。BigRNA从许多个体的配对基因型和128bp分辨率的RNA表达数据中学习,并可以应用于一系列下游任务,如预测RNA结合蛋白(RBP)的特异性和microRNA结合位点。由于BigRNA直接建模RNA-seq数据,它可以发现多种致病的非编码机制,并可以确定它们对转录本的影响。作者展示了BigRNA可以发现非编码变异对表达和剪接的影响,并在恢复已知的致病变异方面达到或超过了专门模型的性能。BigRNA还可以帮助设计不同类型的基于RNA的治疗方法,包括立体阻塞寡核苷酸(SBOs)。在没有额外训练的情况下,BigRNA可以准确识别诱导特定剪接变化的化合物,并高度特异地恢复已知的批准SBO治疗方法。BigRNA理解调控机制的能力还使其能够设计阻断预测的抑制性区域以增加疾病基因表达的SBOs。BigRNA代表了新一代的大规模深度学习模型,可应用于各种不同的个性化RNA治疗发现任务。
BigRNA准确预测了组织特异性的RNA表达以及蛋白质和microRNA的结合位点
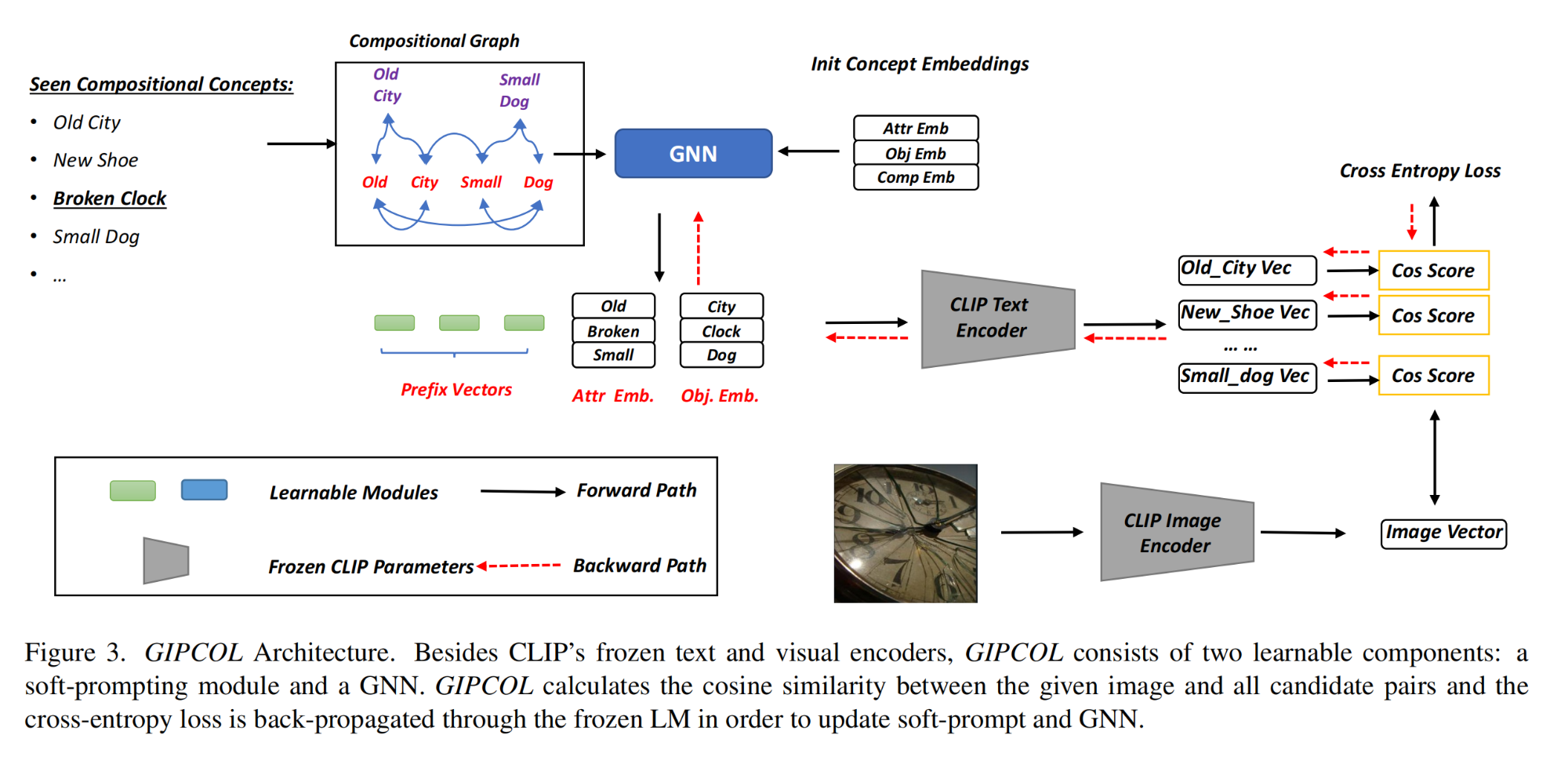
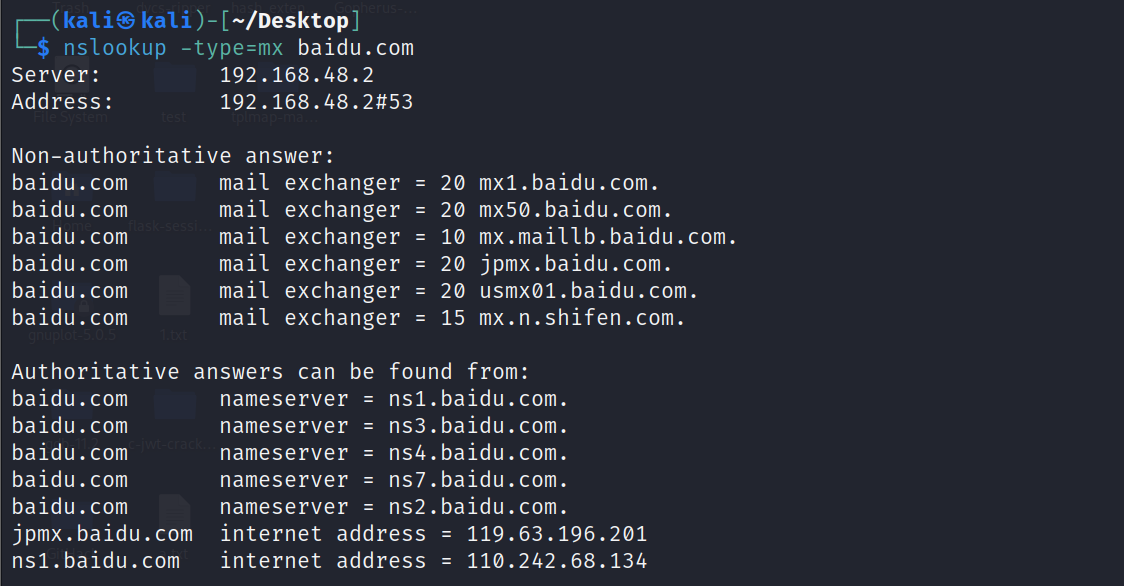
为了训练BigRNA以从相应的DNA序列预测RNA-seq数据,作者采用了基于Transformer的架构,并利用了GTEx资源。对于一个个体的基因型,作者将两个潜在的单倍型独立输入到模型的相同实例中,并训练它以预测这些单倍型的组合输出所观察到的RNA-seq数据(图1a)。模型的每个输出“头”预测一个GTEx样本的表达,因此它学会了预测来自70个个体的2,956个RNA-seq样本在总共51种组织中的输出。在这些RNA-seq数据集上训练后,模型被微调以预测RBP和microRNA结合位点的特异性。作者首先评估了BigRNA预测未见基因组序列表达的能力。作者测量了模型预测训练集之外基因的组织特异性表达水平的能力。BigRNA在预测未见基因的表达水平方面表现出强大的性能,在所有组织中的相关系数(r)在0.47到0.77之间(平均=0.70,图1b)。作者观察到在脑组织中的性能略高于非脑组织(平均r=0.74对0.69,p=5e-03),并强调该模型能够准确预测下丘脑中的表达水平(r=0.74,图1c)。BigRNA预测了SLC7A8的表达水平,这是测试集中的一种氨基酸转运蛋白,展示了预测总体表达水平和准确描绘内外含子结合点的能力(图1d)。为了评估BigRNA在预测不同组织之间的差异这一更困难的任务上的性能,作者使用BigRNA的预测来计算组织对之间总外显子覆盖度的折叠变化,并将其与观察到的折叠变化进行比较。在所有组织间的比较中,观察到平均相关系数r=0.4,这是因为这项任务更加困难(图1e)。作者强调了肝脏和下丘脑之间的比较(r=0.58,p=7e-64,图1f)以说明这一能力。
由于药物发现任务受益于机制的清晰性,接下来作者研究了经过微调的BigRNA模型在预测RBP结合特异性和microRNA结合位点方面的表现。对于RBP任务,作者使用了一个大规模的资源,包括覆盖150个不同人类RBP的223个数据集的全转录组结合概况,这些数据集来自K562和HepG2细胞。发现BigRNA对许多RBP实现了高平均精度,并在所有与先前发布的DeepRiPe系统共有的142个数据集中表现更好(图1g)。在预测microRNA结合位点时,BigRNA实现了0.84的中位AUC,并在测试的所有12个细胞系中表现优于先前发布的方法TargetScan(图1h)。
预测变异对基因表达的影响
在人类遗传学中的一个关键挑战是预测可能存在于人类群体中的序列变异的影响。许多在使用某些指标时对未见过的基因表现良好的深度学习模型,如AlphaFold,很难预测变异的影响。虽然一些准确的方法用于预测罕见的错义变异的致病影响,但非编码变异,例如位于基因的3'和5'未翻译区域(UTR)内的变异仍然难以解释。
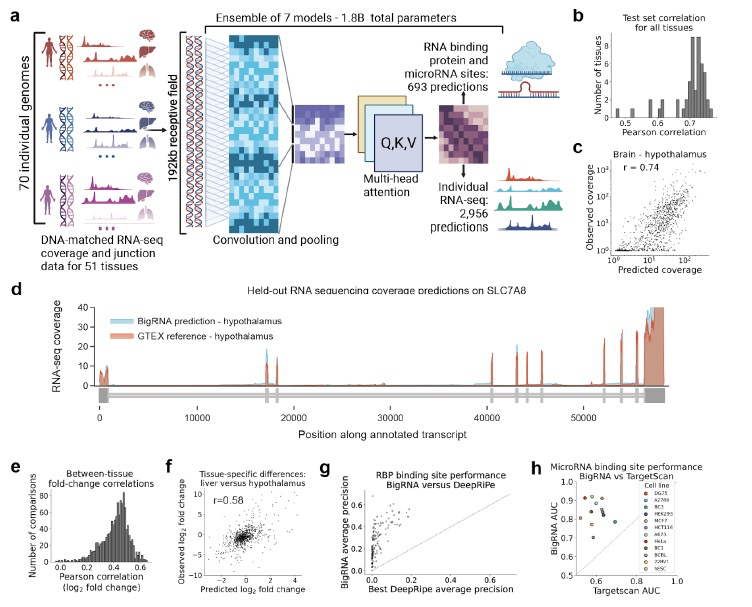
为了填补这一空白,作者评估了BigRNA预测ClinVar中的一组经过筛选的致病性或可能致病性(P/LP)UTR变异的影响能力。作者发现,BigRNA在预测这些变异对其相关疾病基因表达的影响时表现出了强大的性能,无论是在3'UTR还是5'UTR中(AUC=0.95和0.8,图2a)。在5'UTR中的较弱性能可能是由于P/LP变异中调节RNA表达的比例较小,以及影响翻译的机制比例较大(29/47)。作者进一步研究了3'UTR中已知的致病性表达降低变异NAA10。这个变异已知会导致X连锁性显性小眼综合症,并通过破坏NAA10转录物的多腺苷酸化位点(PAS)来降低表达。BigRNA的预测突出了这个变异的表达降低效应(假阳性率FPR <0.5%),并预测了在患者RNA-seq样本中观察到的3'UTR的延长效应(图2b)。对这个变异附近的体外饱和突变分析突出了PAS的重要性,并确认了其他两个附近的P/LP变异的影响(图2c)。
在研究中,作者将BigRNA与其他模型进行了比较,包括Framepool、Saluki和Enformer。与Enformer相比,BigRNA在5'UTR和3'UTR的致病性变异分类方面表现更好。Framepool对5'UTR的致病性变异分类表现与BigRNA类似,但BigRNA在分类已知调节RNA表达的一部分致病性5'UTR变异方面表现更好。
在这些基因中,作者注意到它们的未翻译区域存在许多不确定意义(VUS)的变异。将BigRNA应用于这些变异,以5%的假阳性率(FPR)阈值为基础,在3'UTR中找到了12个潜在的表达调控变异,在5'UTR中找到了23个(图2d)。例如,HBB的3'UTR具有超过此阈值的最高数量的VUS(n=6)。得分最高的VUS位于该基因的PAS中,与已知的致病变异具有相同的位置。HBB的PAS区域还包含大多数已知的P/LP变异(8个中的6个)。第二高得分的VUS(c.*47C>G)位于PAS之外,关于其功能的了解较少。进一步研究发现,尽管被分类为VUS,但此变异据报道会导致HBB的表达下降,支持了BigRNA的预测。还注意到HBB PAS中的其他三个P/LP变异,由于在ClinVar提交中缺乏证据,因此未包含在基准测试中,但它们的得分超过了此阈值,为它们的P/LP分类提供了计算支持。
在更复杂的遗传疾病中,由于连锁不平衡(LD),发现引起表达调控的病因性变异可能具有挑战性。例如,在GTEx中,rs705379和rs854572都被注释为Paraoxonase 1(PON1)的表达性状定量位点(eQTLs),但荧光素酶报告基因检测和该位点的统计映射显示,只有rs705379对表达产生影响,这与BigRNA的预测效果和方向一致,尽管存在较强的LD。BigRNA还为另外两个已知的表达调控变异rs854571和rs3735590分配了更强的效应(图2f),并且正确预测了方向。为了更广泛地评估BigRNA,作者评估了其识别经精细映射的eQTLs的能力,这些eQTLs与效应基因(eGene)、距离转录起始位点(TSS)和次等位基因频率匹配的负对照相对应。作者在这项任务中看到了相当好的性能(AUC = 0.74,图2g),并且相对于Enformer有所提高。
预测变异对基因剪接和内含子保留的影响
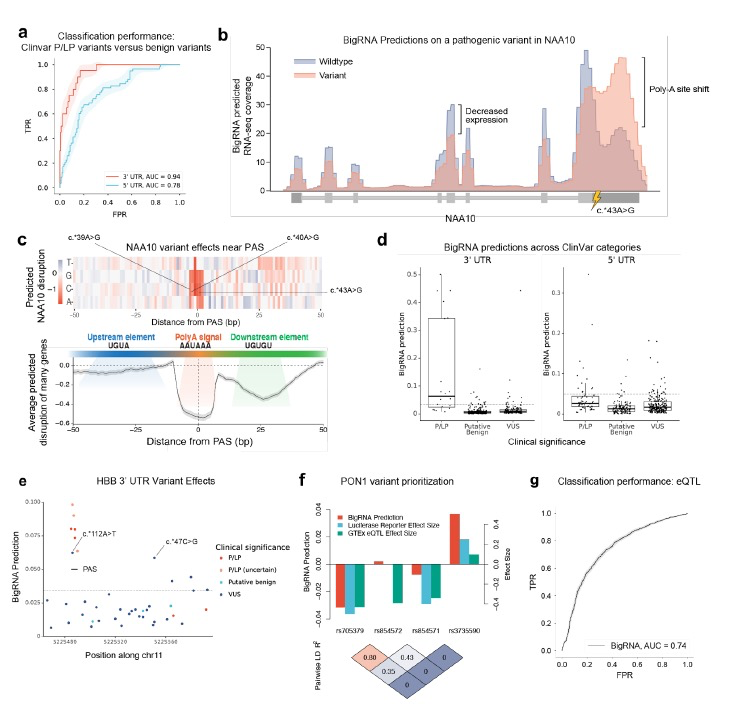
一类重要的病因性变异影响了基因剪接,例如那些导致外显子跳跃的变异。这些变异通常发生在编码区域,并且可能基于其氨基酸替代而被错误地分类为良性突变,尽管它们具有病因性的剪接效应。作者评估了BigRNA对外显子变异的剪接影响的分类能力,这些变异导致大幅度(>50%)的外显子跳跃,与那些不引起任何剪接变化的变异相比,使用了大规模并行剪接分析(MaPSy)的结果。通过预测这些变异引起的连接覆盖变化,BigRNA能够准确地预测这些跳跃变异(AUC = 0.89,图3a),并在这项任务上表现比先前发表的方法SpliceAI 更好(AUC=0.80,差异小于1e-05)。作者进一步研究了一个引起ACADM基因外显子6跳跃的病因性变异,可能导致潜在的中链酰CoA脱氢酶缺乏症。BigRNA预测了这个变异的外显子跳跃效应(FPR = 0.002,图3b),并且它通过创建TDP-43蛋白的结合位点来引起这种跳跃,从而提供了关于作用机制的见解。作者进一步研究了ATP7B中的一个VUS,该基因清除肝细胞中的铜,当其缺陷时会导致威尔逊氏病。BigRNA预测这个变异会导致ATP7B外显子14的跳跃(FPR=0.004,图3c),该外显子包含ATP位点和其他关键元素,因此导致了病因性的功能丧失。
另一类病因性剪接变异是引起完整内含子保留的隐蔽剪接突变。作者评估了BigRNA对一组已报道的内含子保留变异的分类能力,使用附近的常见变异作为负对照。观察到在分类这些变异方面表现出很强的性能(AUC=0.9,图3d),因此进一步研究了BigRNA是否能够预测更复杂的剪接异常。作者将注意力集中在ABCA4基因中的一种病因性非典型剪接位点变异(c.5714+5G>A),已发现它通过引起ABCA4外显子40的跳跃而诱发Stargardt病 。这个变异被强烈预测会引起外显子40的跳跃和内含子40的保留(FPR=0.008和<0.04,图3e),但后者尚未被报告,可能是由于实验技术的限制 。
设计剪接切换和表达增加分子
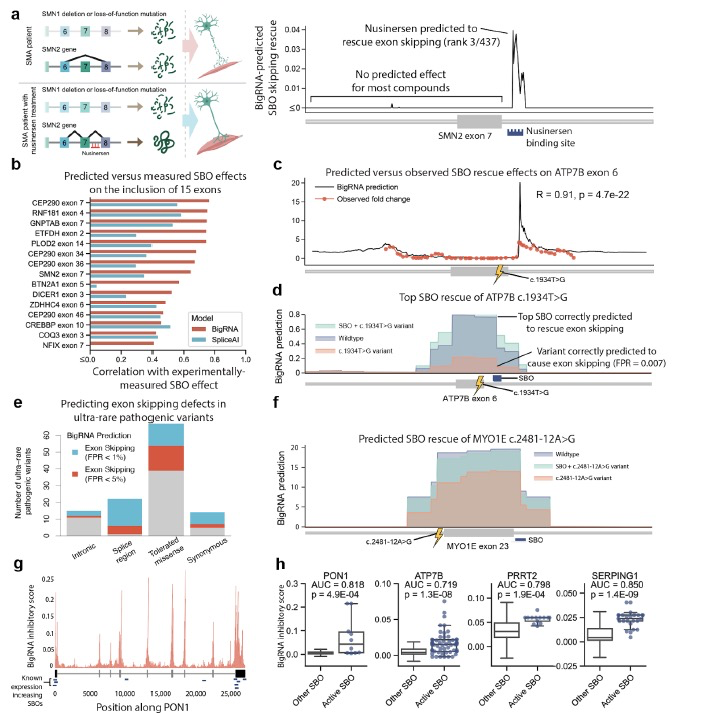
BigRNA的能力在理解影响剪接和基因表达的调控机制方面可能使其能够设计治疗干预措施,以挽救致病变异的影响。在这个应用中,作者评估了BigRNA是否能够通过设计靶向特定RNA的短、化学修饰的合成核酸链(称为"steric blocking oligonucleotides"或SBOs)来逆转剪接缺陷,以调控剪接和基因表达。例如,经FDA批准的Nusinersen是一种SBO,通过逆转SMN2基因中的外显子7跳跃,从而恢复SMN蛋白水平,减轻运动神经元损失和肌肉萎缩,治疗脊髓性肌肉萎缩症。预测SBO的效果的一种方法是隐藏模型输入中的互补结合位点。这种方法属于“零样本学习”的一个示例,因为在进行预测时没有使用任何额外的任务特定的SBO数据。为了评估零样本学习在虚拟筛选中的实用性,作者首先评估了BigRNA在SMN2外显子7的200碱基对内重新发现Nusinersen的能力。令人惊讶的是,BigRNA在437个化合物中将Nusinersen排名前三(图4a)。为了更系统地评估这种方法的有效性,作者对12个基因中的15个外显子进行了总共620个SBO的处理,并观察到在所有情况下预测和实验测得的外显子包含水平之间存在强有力且统计学显著的相关性(r=0.41-0.77,p=7e-12到2e-2,图4b)。
然后,作者使用BigRNA设计了一种新的剪接切换SBO,以挽救一种致病的剪接缺陷。此前,作者曾报道了ATP7B基因中的一种错义变异(c.1934T>G,Met645Arg)通过促使外显子6的跳跃来引发威尔逊病,从而导致功能性蛋白质水平降低,并随后在肝细胞中累积铜。作者在HepG2细胞中创建了Met645Arg变异的疾病模型,并使用这个系统测试了一组靶向被跳跃外显子的SBOs。作者观察到预测的剪接变化与实际测量的剪接变化之间存在强有力的关系(r=0.91,p=4.7e-22,图4c)。这项实验中排名最高的化合物被BigRNA预测为在458种可能的化合物中排名前7。总之,BigRNA预测了Met645Arg引起的外显子跳跃(FPR=0.007)以及最高的实验证明化合物的恢复效应(图4d)。
参考资料
Celaj, A., Gao, A. J., Lau, T. T., Holgersen, E. M., Lo, A., Lodaya, V., ... & Frey, B. J. (2023). An RNA foundation model enables discovery of disease mechanisms and candidate therapeutics. bioRxiv, 2023-09.