文章目录
- 简介
- 安装
- 使用
- Many to Man-使用基因组路径作为输入
- One to One
- 结果
- 其他参数说明
- 可视化两个基因组之间的保守区域
- 并行化
简介
FastANI 是为快速计算全基因组平均核苷酸同一性(Average Nucleotide Identity,ANI)而开发的,无需比对。ANI 的定义是两个微生物基因组之间共享的同源基因对的平均核苷酸同一性。FastANI 支持对完整基因组和基因组草图进行成对比较。其基本程序与 Goris 等人 2007 年描述的工作流程相似。不过,它避免了费时的序列比对,而是使用 Mashmap 作为基于 MinHash 的序列映射引擎来计算同源比对和比对同一性估计值。根据我们对完整基因组和基因组草图的实测试,其准确性与基于 BLAST 的 ANI 的计算相当,而且速度提高了两到三个数量级。因此,它适用于大量基因组对的成对 ANI 计算。有关其速度、准确性和潜在应用的更多详情,请参考对 9 万个原核生物基因组的高通量 ANI 分析揭示了清晰的物种界限"(High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries)。
安装
conda install -c bioconda fastani
使用
Many to Man-使用基因组路径作为输入
(gtdbtk) [yutao@myosin Eisenbacteria]$ head fa.path
./GCA_001780165.1_genomic.fa
./GCA_003235575.1_genomic.fa
./GCA_005893165.1_genomic.fa.gz
(gtdbtk) [yutao@myosin Eisenbacteria]$ time fastANI --ql fa.path --rl fa.path -o ANI.txt --matrix --visualize -t 30
# -ql query list
# -rl reference list
# -o 成对ANI计算结果
# -matrix 同时输出矩阵,以-o选项后的名称加上".matrix"
# --visualize 可视化
# -t thread
One to One
计算单个查询基因组和单个参考基因组之间的 ANI
$ ./fastANI -q [QUERY_GENOME] -r [REFERENCE_GENOME] -o [OUTPUT_FILE]
结果
在上述所有用例中,OUTPUT_FILE 将包含以制表符分隔的行,其中有查询基因组、参考基因组、ANI 值、双向片段比对计数和查询片段总数。比对分数(相对于查询基因组)是映射和片段总数的简单比率。用户还可以选择通过提供 --matrix 参数获得第二个 .matrix 文件,其中包含以 phylip 格式的下三角矩阵排列的标识值。**注意:如果 ANI 值远低于 80%,则不会报告基因组对的 ANI 输出。**这种情况应在氨基酸水平上计算,参见CompareM-平均氨基酸一致性(AAI)计算。
- 示例1
(gtdbtk) [yutao@myosin Eisenbacteria]$ head ANI.txt ANI.txt.matrix
==> ANI.txt <==
# genomeA genomeB ANI(%)
GCA_001780165.1_genomic.fa GCA_001780165.1_genomic.fa 100 1170 1176
GCA_001780165.1_genomic.fa GCA_005893365.1_genomic.fa 78.8657 431 1176
GCA_001780165.1_genomic.fa GCA_005893225.1_genomic.fa 78.8112 381 1176
GCA_001780165.1_genomic.fa GCA_011357805.1_genomic.fa 78.7589 529 1176
GCA_001780165.1_genomic.fa GCA_005893185.1_genomic.fa 78.4959 313 1176
GCA_001780165.1_genomic.fa GCA_005893295.1_genomic.fa 78.2516 308 1176
GCA_001780165.1_genomic.fa GCA_013140805.1_genomic.fa 77.7736 309 1176
GCA_001780165.1_genomic.fa GCA_903921835.1_genomic.fa 77.3711 302 1176
GCA_001780165.1_genomic.fa GCA_902826705.1_genomic.fa 77.3634 256 1176
GCA_003235575.1_genomic.fa GCA_003235575.1_genomic.fa 100 881 888
==> ANI.txt.matrix <==
17
GCA_001780165.1_genomic.fa
GCA_003235575.1_genomic.fa NA
GCA_005893165.1_genomic.fa NA 77.231445
GCA_005893185.1_genomic.fa 78.464157 NA NA
GCA_005893225.1_genomic.fa 78.779877 NA NA 78.797363
GCA_005893265.1_genomic.fa NA NA 79.801826 NA NA
GCA_005893275.1_genomic.fa NA NA 79.954613 NA NA 84.944542
GCA_005893295.1_genomic.fa 78.282249 NA NA 78.198181 78.400757 NA NA
GCA_005893305.1_genomic.fa NA 77.308350 77.910553 NA NA 77.764481 77.908798 NA
- 示例2
(gtdbtk) [yutao@myosin Krumholzibacteriota]$ head Krumholzibacteriota_ANI.txt Krumholzibacteriota_ANI.txt.matrix
==> Krumholzibacteriota_ANI.txt <==
GCA_002085285.1_genomic.fa GCA_002085285.1_genomic.fa 100 425 435
GCA_002403075.1_genomic.fa GCA_002403075.1_genomic.fa 100 772 776
GCA_002403075.1_genomic.fa GCA_002403295.1_genomic.fa 86.9047 500 776
GCA_002403295.1_genomic.fa GCA_002403295.1_genomic.fa 99.9999 599 615
GCA_002403295.1_genomic.fa GCA_002403075.1_genomic.fa 86.8991 506 615
GCA_002747875.1_genomic.fa GCA_002747875.1_genomic.fa 100 936 948
GCA_002747875.1_genomic.fa GCA_002790835.1_genomic.fa 78.4217 265 948
GCA_002747875.1_genomic.fa GCA_903847545.1_genomic.fa 78.1095 189 948
GCA_002747875.1_genomic.fa GCA_003646045.1_genomic.fa 78.0435 186 948
GCA_002747875.1_genomic.fa GCA_903859215.1_genomic.fa 77.8668 240 948
==> Krumholzibacteriota_ANI.txt.matrix <==
16
GCA_002085285.1_genomic.fa
GCA_002403075.1_genomic.fa NA
GCA_002403295.1_genomic.fa NA 86.901932
GCA_002747875.1_genomic.fa NA NA NA
GCA_002790835.1_genomic.fa NA NA NA 78.286392
GCA_003353795.1_genomic.fa NA NA NA NA 76.844803
GCA_003369455.1_genomic.fa NA NA NA NA NA NA
GCA_003369535.1_genomic.fa NA NA NA NA NA NA 77.283585
GCA_003646045.1_genomic.fa NA NA NA 78.091743 78.153748 77.447433 NA NA
其他参数说明
# version 1.32
-r <value>, --ref <value>
reference genome (fasta/fastq)[.gz]
--refList <value>, --rl <value>
a file containing list of reference genome files, one genome per line
-q <value>, --query <value>
query genome (fasta/fastq)[.gz]
--ql <value>, --queryList <value>
a file containing list of query genome files, one genome per line
-t <value>, --threads <value>
thread count for parallel execution [default : 1]
--visualize
output mappings for visualization, can be enabled for single genome to
single genome comparison only [disabled by default]
--matrix
also output ANI values as lower triangular matrix (format inspired from
phylip). If enabled, you should expect an output file with .matrix
extension [disabled by default]
-o <value>, --output <value> [required]
output file name
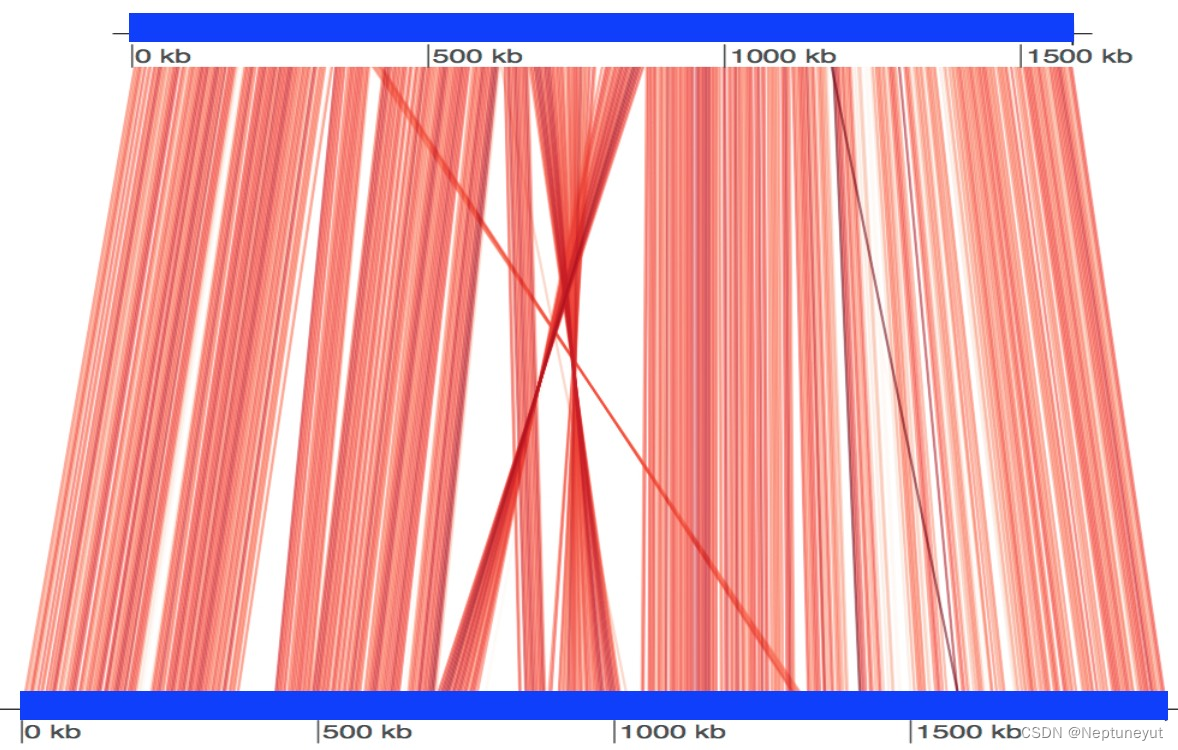
可视化两个基因组之间的保守区域
FastANI 支持将两个基因组之间计算出的比对区域可视化。要获得这种可视化效果,需要如上所述使用 FastANI 进行一对一比较,但需要提供一个额外的标记–visualize。该标志会强制 FastANI 输出一个映射文件(扩展名为 .visual),其中包含所有互易映射的信息。最后,资源库中提供了一个 R 脚本,该脚本使用 genoPlotR 软件包绘制这些映射图。这里我们展示一个使用两个基因组运行的示例:Bartonella quintana(GenBank:CP003784.1)和 Bartonella henselae(NCBI 参考序列:NC_005956.1)。
$ ./fastANI -q B_quintana.fna -r B_henselae.fna --visualize -o fastani.out
$ Rscript scripts/visualize.R B_quintana.fna B_henselae.fna fastani.out.visual
并行化
FastANI(v1.1 及以后版本)支持多线程,使用-t配置线程数。要使 FastANI 的并行化超越单个计算节点,用户还可以选择简单地将参考数据库划分为多个分块,并将它们作为并行进程执行。我们在资源库中提供了一个脚本,用于随机分割数据库。