一、药物理化性质预测
药物的理化性质包括分子的脂溶性、水溶性和解离常数(Dissociation Constant)pKa,这些理化参数与药代动力学性质、生物活性强度以及作用靶标的选择性密切相关,下面分别介绍。
1、脂溶性(logP)
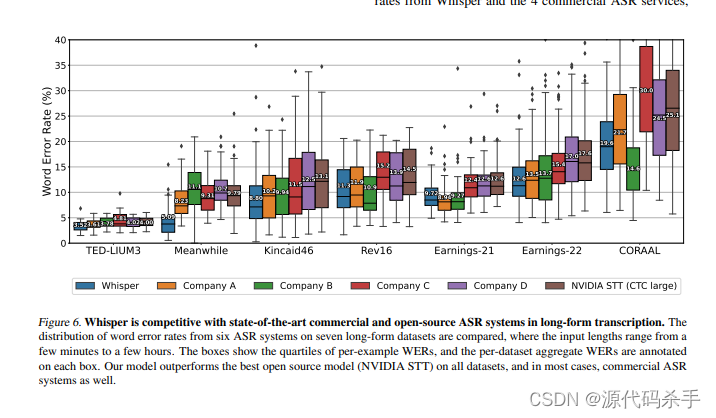
脂溶性(Lipophilicity)是药物发现和设计过程中的一个重要参数,也叫疏水性(Hydrophobicity),与化合物的水溶性、膜渗透性、活性强度、选择性、杂泛性等密切相关,也影响着化合物的药代动力学和药效学性质。适宜的脂溶性大小(Logp)、较低的分子量(MW)和极性表面积(TPSA)是一个化合物具有良好吸收的主要驱动力。
最常用的表征脂溶性的参数是分配系数(Partition Coefficient)logP值。logP 是非离子化的化合物(中性化合物)在水相和有机溶剂相(一般为正辛醇)中的浓度比。药物化学家依据化合物的结构来计算logP值,理想药物的脂溶性logP值一般在1~5之间。在1964年,Fujita和Hansch发展了取代基疏水参数π,用来估算芳香取代物的分配系数【1】。此后,各种不同的方法被相继提出,用来计算化合物logP值。这些计算方法大致可以分为两大类:基于子结构的加和模型法和基于性质的方法。
基于子结构的加和模型法
先把分子分割成片段(Fragment)或单个原子,然后将这些片段或单个原子的疏水贡献进行加和。
基于性质的方法
根据整个分子的理化性质,如分子表面、分子体积、偶极矩、部分电荷、轨道能或者一些拓扑指数以及静电指数等。
2、水溶性(logS)
水溶性(Solubility)是表征化合物类药性的重要参数,也是影响药物吸收和生物利用度的最重要因素之一。药物在体内要被吸收,首先必须溶解于水,然后才有机会透过生物膜。在药物研发早期,由于需要提高化合物的活性,往往引入脂溶性结构来增强与靶标蛋白的亲和力,这使得化合物的水溶性问题变得突出。
较好的水溶性:一个平均亲和力为1 mg/kg的化合物的溶解度至少为100 μg/mL才能称为水溶性较好。
计算水溶性模型主要是基于不同描述符的统计学方法,通过构建QSAR方程来预测水溶性。水溶性常用其对数形式logS表示。常用的水溶性预测方法包括计算量要求比较高的基于量子力学和分子力学的自由能计算方法和速度相对较快的QSAR方法。后一种方法更适合于大规模化合物库的计算,在药物设计中也是应用最多的。
3、pKa值
pKa值用来表征化合物的解离常数(Dissociation Constant),和药物的ADME性质密切相关。药物的分布和扩散严重依赖药物在生理pH值下的离子化状态,药物的中性形式具有更强的脂溶性,而离子化形式则更具极性和水溶性。常见的计算方法一般可分为基于量子力学的方法和基于QSAR的方法。
在新药研发的过程中,大部分化合物会因为缺乏药效、不良药代动力学性质、动物体内毒性、人体副作用或一些商业因素而最终无法成为药物。在这些因素中,药代动力学性质和毒性问题造成的失败率高达50%,因此也成为药物研发过程中重点关注的方向。
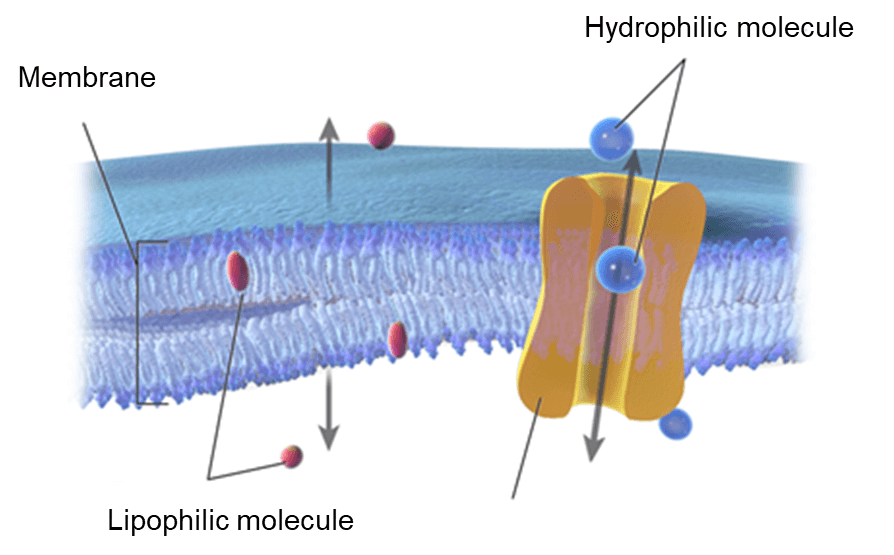
药代动力学(Pharmacokinetics,PK)主要研究体内药物浓度随时间变化的规律,涉及ADME等方面,即药物在体内的吸收(Absorption)、分布(Distribution)、代谢(Metabolism)、和排泄(Excretion)过程。ADME决定了药物在体内的生物利用度、作用时间长短和所需剂量大小。除此之外,毒性(Toxicity)与药代动力学也密切相关,通常与ADME结合起来考虑,即ADME/T。
在药物研发的过程中,不少药物化学家遵循“失败早,损失小”的原则,提出在先导化合物的发现阶段进行ADME/T性质预测,选择性质理想的化合物进行实验筛选,从而降低研发成本,提高候选药物后期开发的成功率。
二、药物的体内过程和药代动力学性质预测
药物由给药部位进入机体产生药效,再由机体排出,其间经历了吸收、分布、代谢和排泄四个过程。下文介绍药物的体内过程、药代动力学性质和预测方法。
1、吸收
药物的吸收(Absorption) 是指药物从用药部位进入血循环的过程。通常使用生物利用度(所给药物进入人体循环的药量比例)来反映吸收过程的速度和程度。
口服药物被吸收后经门静脉进入肝脏,有些药物首次进入肝脏就被肝药酶代谢,进入体循环的药量减少,称为首关消除(First Pass Elimination)。经过肝脏首关消除后,药物随着血液循环分布到全身组织、器官和预期的作用靶标,发生期待的药理作用。
化合物在人体的吸收主要与水溶性和膜渗透性密切相关。水溶性的预测在下一章节中会介绍,膜渗透性主要与脂溶性有关,相关的预测方法包括人体小肠吸收性预测和血脑屏障渗透性预测。
(1)人体小肠吸收性预测
人体小肠吸收(Human Intestinal Absorption,HIA)是口服给药途径中药物转运到靶标的关键步骤,影响药物在体内的生物利用度。药物透过被动扩散(占绝大多数)和载体主动转运的方式被小肠吸收。许多因素会影响药物的小肠渗透,如脂溶性、氢键形成能力、分子大小等。
HIA是药物研发早期需要考虑的重要药代动力学性质之一。计算预测HIA的模型分为基于QSAR的定量预测模型和基于各种机器学习方法的定性分类模型。
(2)血脑屏障渗透性预测
血脑屏障(Blood Brain Barrier,BBB)是一种存在于血液与脑组织之间的内皮细胞屏障。BBB可阻止多种物质进入脑内,对于作用于中枢神经系统的药物,需要其具有较高的穿透BBB的能力;对于非作用于中枢神经系统的药物,则要求尽量降低其穿透能力,从而避免对中枢神经系统产生意外的副作用。在药物研发的过程中,化合物的BBB穿透能力是非常重要的一个性质。
计算预测血脑屏障的模型主要包括基于各种描述符的定性分类模型和QSAR定量回归模型。比较这两个模型,定量回归模型只能包含少量化合物样本,定性分类模型可以包含数量更多、结构差异性更大的化合物样本。
2、分布
药物的分布(Distribution ) 是指药物从血循环系统到达组织器官的过程。影响分布的因素包括:
① 药物本身的理化性质(包括分子大小、脂溶性、pKa等);
② 药物与血浆蛋白结合率:结合药不能通过生物膜,只有游离药物才能向组织分布;
③ 组织器官的屏障作用,如血脑屏障、胎盘屏障;
④ 细胞膜两侧体液的pH。与分布相关的ADMET参数包括分布容积、血浆蛋白结合、中枢神经渗透、P-糖蛋白外排。
药物进入血液后,一部分与血浆蛋白结合,一部分以游离的形式存在于血液中,通过细胞膜转运到特定的靶标部位发挥作用,药物在中枢神经中的浓度与药物通过血脑屏障的能力以及转运药物的P-糖蛋白有关,主要的预测模型有血浆蛋白结合模型,中枢神经渗透模型和P-糖蛋白抑制剂和底物预测模型。
(1)P-糖蛋白结合预测模型
P-糖蛋白(P-Glycoprotein,P-gp)是ABC转运体超家族的一员,广泛地分布于多个人体组织中。P-糖蛋白拥有非常宽的底物特异性,能结合包括药物在内的不同类型和大小的化合物。另外,P-糖蛋白具有外排性,会将不同的药物排出癌细胞,导致出现多药耐受现象。P-糖蛋白对于药物的吸收、分布和毒性起着极其重要的作用。
计算预测P-糖蛋白的模型主要包括P-糖蛋白底物预测模型和P-糖蛋白抑制剂预测模型。
(2)化合物血浆蛋白结合率预测模型
化合物的血浆蛋白结合率(Plasma Protein Binding,PPB)是一个重要的药代动力学参数,影响着药物在体内的分布和清除。结合型药物(指与血浆蛋白结合的药物)会随着血液循环到达全身;非结合型药物(或称游离药物)能够进入组织和相应的靶标蛋白结合,产生相应的药理学效应。PPB还与药物的相互作用有关,当同时使用两个与血浆蛋白都有高结合的药物时,竞争结合会导致药物结合率下降,游离浓度升高,引起严重的不良反应。
计算预测PPB的模型主要包括运用非线性回归分析法、机器学习方法和定量回归模型。
(3)小分子亚细胞定位预测模型
药物在人体细胞中会被定位到不同的亚细胞器中,不同的亚细胞器有不同的功能和微环境,药物在其中的浓度也不一样。
如果小分子能够定位到特定的亚细胞器中,则更容易发挥药效;相反,若其富集在其它亚细胞器中,则可能导致毒副作用。因此,在药物设计中通过研究化合物在亚细胞中的定位情况,能够增加药物与靶蛋白结合的可能性,并减少潜在的毒副作用。
计算预测小分子的亚细胞定位的方法模型包括依据已有的小分子性质与其定位关系使用统计学回归的QSAR方法建立模型和基于机制的生理学模型。
3、代谢
药物的生物转化(Biotransformation)又称代谢(Drug Metabolism),是指药物的化学结构在体内药物代谢酶的作用下发生改变的过程。药物的代谢性质关系到药物的生物利用度、化学稳定性和安全性。
计算预测药物的技术可以分为基于配体的方法和基于结构的方法。
(1)代谢位点预测
在一个分子中,代谢反应发生的位点被称为代谢位点(Site of Metabolism,SOM)。代谢位点的预测对提高药物的生物利用度和避免产生可能的有毒产物非常重要。计算预测代谢位点的方法大体分为:基于配体的M预测、基于规则的SOM预测、整合基于配体和基于结构的综合SOM预测。
(2)代谢产物预测
知道一个小分子的代谢产物结构,有助于理解药代动力学性质和进行药物设计。预测代谢产物的难点在于同时正确识别发生代谢反应的位点和代谢反应的类型。目前大部分预测代谢产物的方法都是基于知识的系统,即专家系统。
(3)P450酶抑制预测
临床上对患者同时给药是一种常见现象,如果其中一种药物在体内产生P450酶抑制,就会改变其代谢能力,从而改变其他药物的药效或引起药物安全性问题。常见的预测方法分为针对大量化合物的抑制剂分类预测和基于某一特定P450酶亚型进行抑制剂的筛选预测。
4、排泄
排泄(Excretion)是药物以原形或代谢产物的形式经不同途径排出体外的过程。药物及其代谢产物经肾脏从尿液排泄,其次经胆汁从粪便排泄。挥发性药物主要经肺随呼出气体排泄。除此之外,药物也可经汗液和乳汁排泄。
三、代谢药物毒性预测模型
急性毒性 (Acute Toxicity)是指机体因一次或在24小时内多次接触外源化学物而产生的中毒效应或死亡,这是药物安全性评价的一个重要指标。
1、口服急性毒性预测
药物的急性毒性可以分为口服、经皮和吸入性急性毒性。由于药物主要以口服为主,以此构建的预测模型是研发过程中的重点研究对象。现有的预测模型包括局部模型(Local Model)和全局模型(Global Model)两类。
2、致突变性预测
致突变性(Mutagenicity)是指化合物引起细胞核中的DNA发生改变。由于这种改变会随细胞的分裂过程而传递,因此化合物致突变性是药物安全性的重要指标。
致突变物(Mutagen)是指能引起DNA突变的物质,又称诱变剂。Bruce Ames在1983年提出突变物测试方法(Ames试验),并检测出175已知致癌物。
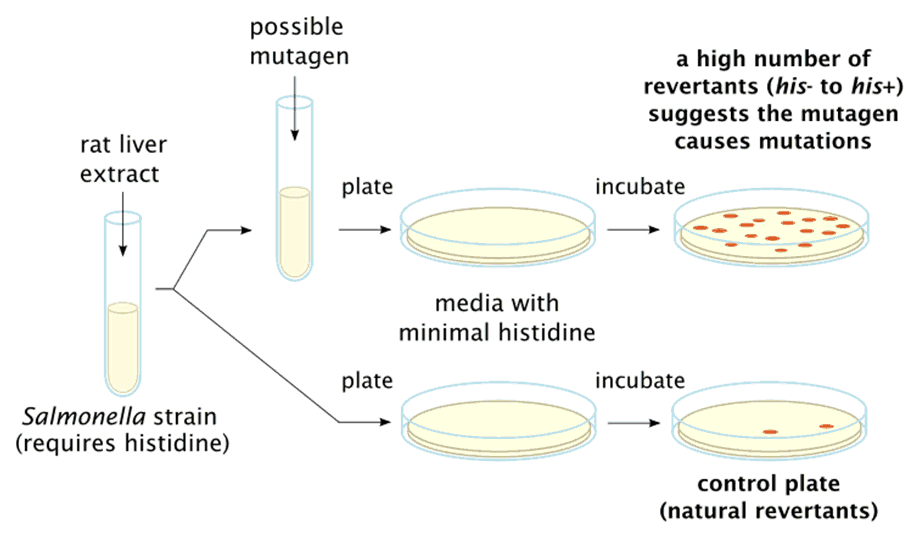
Ames试验流程
其中,超过90%的致癌物具有致突变性。Ames试验通常被做为先期测试方法,并作为模式系统被广泛用于化学物的毒性及致突变性安全评估。
目前,在计算预测领域已有多种致突变性预测模型。依据经验和预测结果分析,把专家系统和基于统计的方法综合起来运用可以获得较好的预测能力。
3、致癌性预测
致癌性(Carcinogenicity)是药物安全性评价的重要指标之一。拥有致癌性的化合物会引发肿瘤或增加肿瘤发生率,严重威胁人类健康,需要尽早进行评估。
化合物致癌性的预测模型也可以分为局部模型和全局模型。局部模型主要集中于N-亚硝基化合物、芳香胺类化合物和多环芳烃化合物等同系物。
4、hERG毒性预测
hERG(the human Ether-a-go-go-related Gene)是人延迟整流钾离子通道基因,该基因编码的蛋白质钾离子通道在动作电位的复极化过程中起着重要的作用。
很多药物由于作用于hERG钾离子通道导致心脏QT间期增长,而造成严重的毒副作用甚至造成猝死。若干临床上成功的上市药物有抑制hERG的趋势,导致其在使用过程中伴随着猝死的药物不良反应风险。比如说特芬那定(Terfenadine)和西沙必利(Cisapride)因为导致hERG相关的心脏毒性而被撤市。因此,在药物研发过程中要尽量避免药物存在hERG抑制。
预测化合物的hERG毒性主要有基于配体和基于受体的两类方法。早期的研究主要是利用药效团和包括CoMFA和CoMSIA在内的3D-QSAR方法建立预测模型,近年来随着机器学习算法的不断发展,各种基于机器学习的hERG毒性分类预测模型涌现而出。
由于hERG-药物小分子复合物的三维结构还没有解析出来,因此基于受体的预测模型主要是组合运用分子对接和分子动力学模拟研究抑制剂和hERG的相互作用,以理解阻断剂阻断hERG活性的机理。但基于受体的预测模型目前在应用上还有不少局限性。
5、药物肝毒性预测(DILI)
药物诱导肝损伤(Drug-Induced Liver Injury, DILI)引起的药物肝毒性(Hepatotoxicity)是常见的药物不良反应。DILI不仅严重危害用药者的健康,还是药物研发失败、限制使用、撤市的主要原因。由于药物诱导肝毒性的分子机制非常复杂,使得对DILI的预测十分困难。随着现代生命科学、计算机、生物信息学等技术的发展,基于药物分子结构的计算模型被广泛地应用于药物引起的肝毒性预测,比如说利用贝叶斯方法、深度学习技术、子结构模式识别方法。这些计算模型在预测肝毒性的实际应用过程中获得了不错的结果。
6、药物内分泌干扰性预测(EDC)
内分泌干扰物(Edocrine-Disrupting Chemical,EDC)是指环境中能够干扰人体内分泌系统,进而对生殖、发育、神经和免疫系统有不良影响的化合物。早期,针对EDC的预测模型多是基于统计学算法和分子描述符建立的,它们中的大部分预测模型需要有限的体外数据支持。近来,机器学习方法脱颖而出,它可以对分子结构进行前瞻性预测,为化合物提供一个更加综合且可靠的内分泌干扰特性预测,且拥有更低的计算成本【3】。

https://dx.doi.org/10.1021/acs.est.0c03982
四、警示子结构识别
警示子结构(Structural Alerts,SA)是指导致化合物产生毒性的结构片段。该片段可以是一个特定的子结构、一个类似Markush结构的结构通式,或者是一些子结构的组合。一个毒性端点可能具有多个警示子结构(即不同的毒性机理),一个警示子结构也可能与多个毒性端点相关,如一些活性官能团能产生细胞毒性,损害不同器官。药物化学家通过警示子结构,可以快速识别潜在的毒性化合物,并对其进行结构优化降低毒性。
1、基于片段的方法
利用化学信息学工具将分子切割成所有可能的片段,并计算各片段出现的频率,如SARpy和CASE。
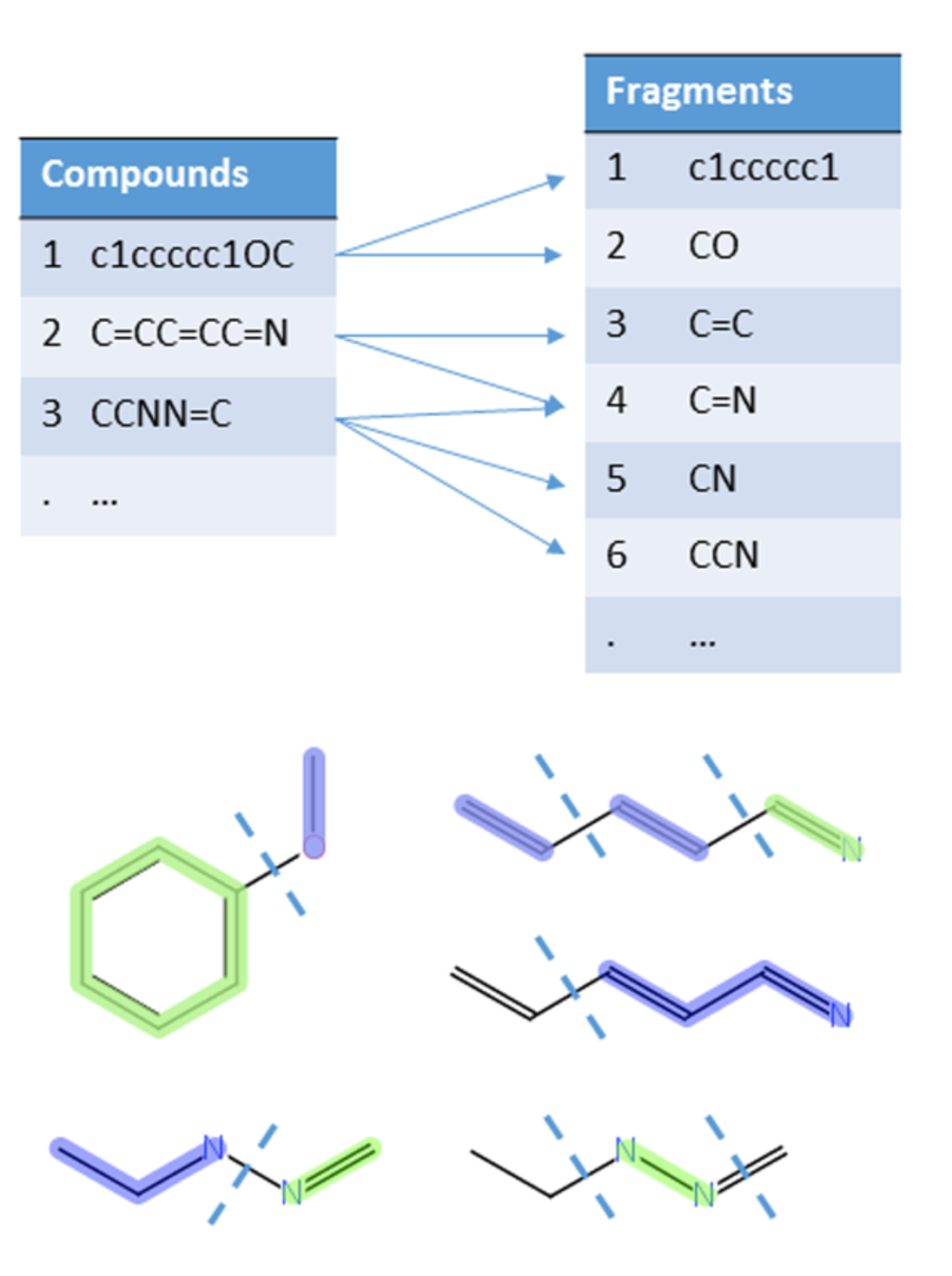
DOI: 10.1021/acs.chemrestox.7b00083
2、基于图的方法
将化合物看作由点和边构成的图,然后应用图论算法进行处理,如MolFea、MoSS和Gaston。
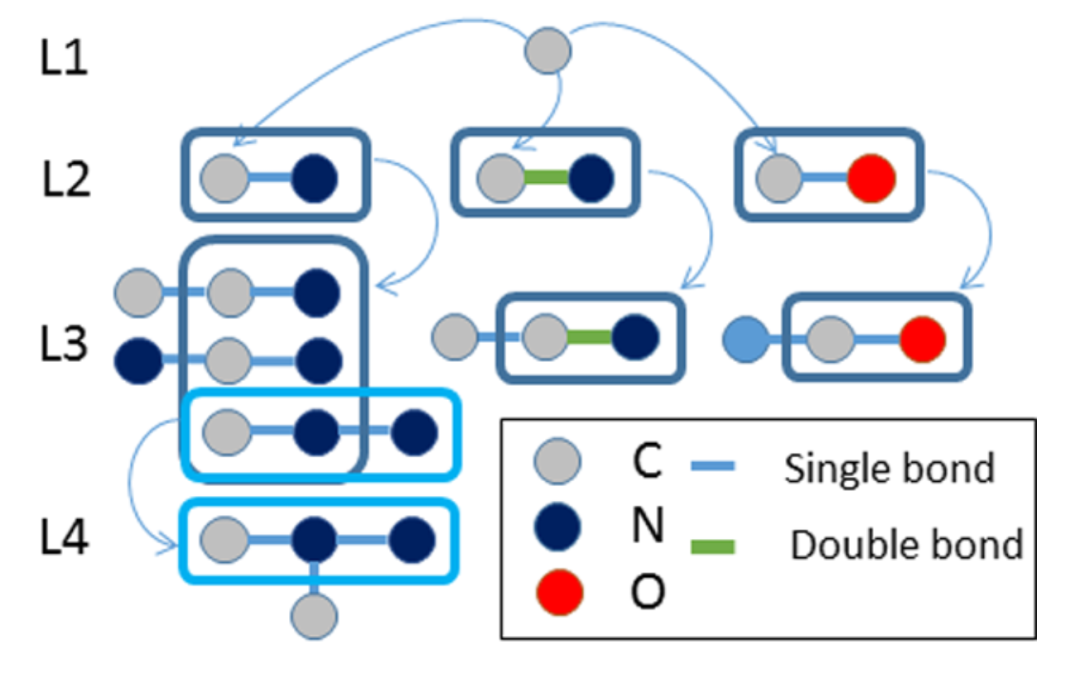
DOI: 10.1021/acs.chemrestox.7b00083
3、基于指纹的方法
采用预先定义的子结构库,也属于特殊的基于片段的方法,如Bioalerts和MACCS。
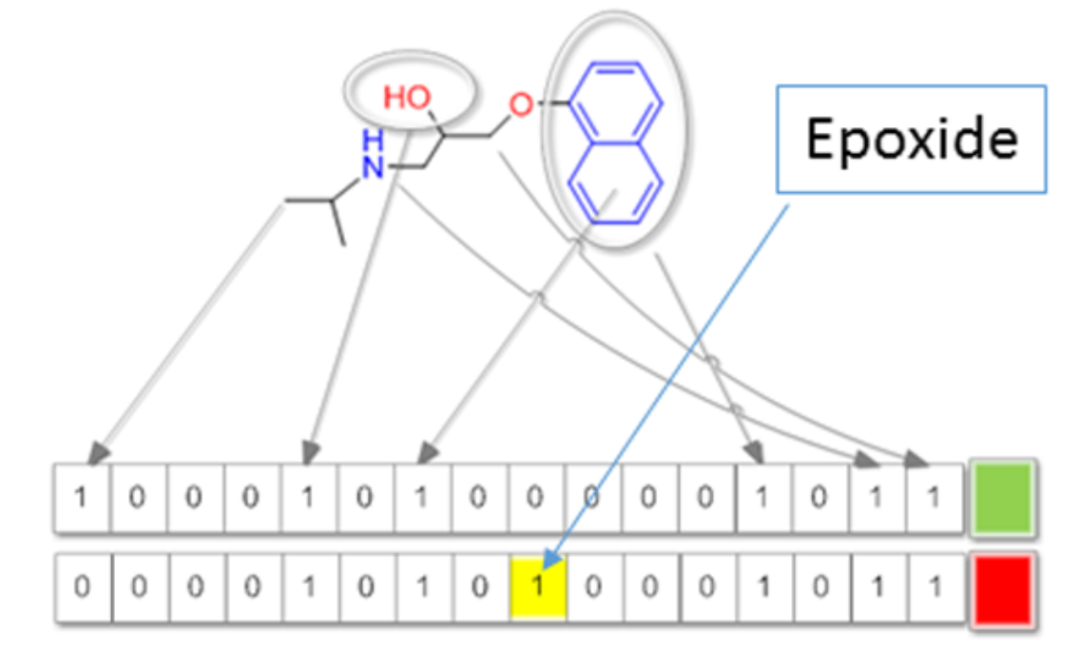
DOI: 10.1021/acs.chemrestox.7b00083
警示子结构可用于快速识别化合物是否具有某种潜在毒性。如果一个分子中出现某个警示子结构,则提示该分子很可能具有某种毒性,药物化学家在研究时需要特别注意,并构建警示子结构库,用于指导未来的药物设计工作。在药物研发后期,发现候选药物具有某种毒性,可以考虑识别其警示子结构,然后对警示子结构进行改造优化,降低其毒性。
优化警示子结构方法
(1)保持原结构不变,只是在警示子结构周围合适的位置增加一个基团,以逆转其毒性。

DOI: 10.1021/acs.chemrestox.7b00083
2)采用片段替换(骨架跃迁)方法,将警示子结构替换为一个无毒性的子结构。
当前警示子结构识别方法存在的问题
(1)识别的准确度还有待于提高,尤其是不同的软件方法得到的警示子结构不同,这需要用户的经验积累;
(2)目前的方法仅限于识别单个警示子结构,尚未考虑多个子结构共存于同一分子内时的相互影响,比如协同效应、拮抗效应。
万字长文 | 药物的吸收、分布、代谢和排泄(药代动力学)
跟我学药物设计 | 药代动力学性质与毒性预测








![[Hive] explode](https://img-blog.csdnimg.cn/89a7d7071c1345e6a89c9118c1d4edb9.png)