目录
一、实验介绍
二、实验环境
1. 配置虚拟环境
2. 库版本介绍
3. IDE
三、实验内容
0. 导入必要的工具包
1. 定义一些阈值和参数
2. 读取数据
normal_data.csv部分展示
tumor_data.csv部分展示
3. 绘制箱型图
4. 删除表达量低于阈值的基因
5. 计算差异显著的基因
6. 初始化结果数据表格
7. 进行秩和检验和差异倍数计算
8. 可视化分析
9. 代码整合
一、实验介绍
本实验完成了基因差异分析,包括数据读取、数据处理( 绘制箱型图、删除表达量低于阈值的基因、计算差异显著的基因)、差异分析(进行秩和检验和差异倍数计算)等,成功识别出在正常样本与肿瘤样本之间显著表达差异的基因,并对其进行了进一步的可视化分析(箱型图、差异倍数fold分布图、热力图和散点图)。
基因差异分析是研究不同条件下基因表达差异的重要手段,能够帮助我们理解生物体内基因调控的变化及其与表型特征的关联。本实验旨在探索正常样本与肿瘤样本之间基因表达的差异,并识别差异显著的基因。
二、实验环境
本系列实验使用了PyTorch深度学习框架,相关操作如下(基于深度学习系列文章的环境):
1. 配置虚拟环境
深度学习系列文章的环境
conda create -n DL python=3.7 conda activate DLpip install torch==1.8.1+cu102 torchvision==0.9.1+cu102 torchaudio==0.8.1 -f https://download.pytorch.org/whl/torch_stable.html
conda install matplotlibconda install scikit-learn新增加
conda install pandasconda install seabornconda install networkxconda install statsmodelspip install pyHSICLasso注:本人的实验环境按照上述顺序安装各种库,若想尝试一起安装(天知道会不会出问题)
2. 库版本介绍
| 软件包 | 本实验版本 | 目前最新版 |
| matplotlib | 3.5.3 | 3.8.0 |
| numpy | 1.21.6 | 1.26.0 |
| python | 3.7.16 | |
| scikit-learn | 0.22.1 | 1.3.0 |
| torch | 1.8.1+cu102 | 2.0.1 |
| torchaudio | 0.8.1 | 2.0.2 |
| torchvision | 0.9.1+cu102 | 0.15.2 |
新增
| networkx | 2.6.3 | 3.1 |
| pandas | 1.2.3 | 2.1.1 |
| pyHSICLasso | 1.4.2 | 1.4.2 |
| seaborn | 0.12.2 | 0.13.0 |
| statsmodels | 0.13.5 | 0.14.0 |
3. IDE
建议使用Pycharm(其中,pyHSICLasso库在VScode出错,尚未找到解决办法……)
win11 安装 Anaconda(2022.10)+pycharm(2022.3/2023.1.4)+配置虚拟环境_QomolangmaH的博客-CSDN博客![]() https://blog.csdn.net/m0_63834988/article/details/128693741
https://blog.csdn.net/m0_63834988/article/details/128693741
三、实验内容
0. 导入必要的工具包
import numpy as np
import pandas as pd
import scipy
import matplotlib.pyplot as plt
import seaborn as sns
from scipy.stats import zscore
from scipy.stats import ranksums
from pyHSICLasso import HSICLasso
from sklearn.preprocessing import LabelEncoder1. 定义一些阈值和参数
p_cutoff = 0.001
FC_cutoff = 3
num_cutoff = 10p_cutoff是判断差异显著性的 p 值阈值。FC_cutoff是判断差异的倍数阈值。num_cutoff是基因表达量筛选的阈值。
2. 读取数据
ndata = pd.read_csv("normal_data.csv", index_col=0, header=0)
tdata = pd.read_csv("tumor_data.csv", index_col=0, header=0)从文件 "normal_data.csv" 和 "tumor_data.csv" 中读取正常样本和肿瘤样本的基因表达数据。
normal_data.csv部分展示
| TCGA-BL-A13J-11A-13R-A10U-07 | TCGA-BT-A20N-11A-11R-A14Y-07 | TCGA-BT-A20Q-11A-11R-A14Y-07 | TCGA-BT-A20R-11A-11R-A16R-07 | TCGA-BT-A20U-11A-11R-A14Y-07 | TCGA-BT-A20W-11A-11R-A14Y-07 | TCGA-BT-A2LA-11A-11R-A18C-07 | TCGA-BT-A2LB-11A-11R-A18C-07 | TCGA-CU-A0YN-11A-11R-A10U-07 | TCGA-CU-A0YR-11A-13R-A10U-07 | TCGA-GC-A3BM-11A-11R-A22U-07 | TCGA-GC-A3WC-11A-11R-A22U-07 | TCGA-GC-A6I3-11A-11R-A31N-07 | TCGA-GD-A2C5-11A-11R-A180-07 | TCGA-GD-A3OP-11A-11R-A220-07 | TCGA-GD-A3OQ-11A-21R-A220-07 | TCGA-K4-A3WV-11A-21R-A22U-07 | TCGA-K4-A54R-11A-11R-A26T-07 | TCGA-K4-A5RI-11A-11R-A28M-07 | |
| ?1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ?2 | 2.5521 | 3.8933 | 5.4594 | 16.4583 | 34.4264 | 5.2219 | 11.346 | 38.256 | 4.3054 | 8.7471 | 21.0084 | 3.1837 | 0 | 6.0128 | 17.7621 | 40.8452 | 8.3088 | 13.7216 | 1.5099 |
| ?3 | 8.5119 | 5.7305 | 10.4417 | 7.681 | 19.062 | 1.3534 | 3.4219 | 25.7514 | 1.4834 | 6.5471 | 9.6515 | 2.6472 | 5.3121 | 10.7337 | 19.364 | 19.138 | 19.5273 | 18.5364 | 14.4093 |
| ?4 | 117.4995 | 103.9108 | 148.8251 | 100.9537 | 162.7054 | 128.9808 | 118.3924 | 124.8423 | 180.5137 | 131.3118 | 127.6985 | 145.6327 | 95.2855 | 119.1427 | 149.7972 | 128.4473 | 142.8571 | 128.644 | 139.1199 |
| ?5 | 619.5833 | 738.4077 | 679.3286 | 786.7036 | 1132.558 | 1032.877 | 1158.65 | 1143.321 | 687.4096 | 690.5882 | 697.5129 | 697.3761 | 1551.793 | 556.2201 | 1018.907 | 646.4851 | 895.4832 | 903.8232 | 836.9674 |
| ?6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ?7 | 128.3422 | 115.4856 | 119.258 | 120.6965 | 120.9302 | 54.7945 | 82.7004 | 91.3729 | 66.5702 | 153.5294 | 259.3317 | 272.8863 | 291.5007 | 94.4976 | 250.6016 | 253.2622 | 313.5504 | 270.6093 | 187.172 |
| ?8 | 0.7376 | 0 | 0 | 0.3957 | 0.7752 | 0.5479 | 0 | 0.4638 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.3332 | 0 | 0 | 0 |
| ?9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.3987 | 0 | 0 | 0 | 0 | 0 |
tumor_data.csv部分展示
| TCGA-BT-A42F-01A-11R-A23W-07 | TCGA-C4-A0EZ-01A-21R-A24X-07 | TCGA-C4-A0F0-01A-12R-A10U-07 | TCGA-C4-A0F1-01A-11R-A034-07 | TCGA-C4-A0F6-01A-11R-A10U-07 | TCGA-C4-A0F7-01A-11R-A084-07 | TCGA-CF-A1HR-01A-11R-A13Y-07 | TCGA-CF-A1HS-01A-11R-A13Y-07 | TCGA-CF-A27C-01A-11R-A16R-07 | TCGA-CF-A3MF-01A-12R-A21D-07 | TCGA-CF-A3MG-01A-11R-A20F-07 | TCGA-CF-A3MH-01A-11R-A20F-07 | TCGA-CF-A3MI-01A-11R-A20F-07 | TCGA-CF-A47S-01A-11R-A23W-07 | TCGA-CF-A47T-01A-11R-A23W-07 | TCGA-CF-A47V-01A-11R-A23W-07 | TCGA-CF-A47W-01A-11R-A23W-07 | TCGA-CF-A47X-01A-31R-A23W-07 | TCGA-CF-A47Y-01A-11R-A23W-07 | TCGA-CF-A5U8-01A-11R-A28M-07 | |
| ?1 | 0 | 0.3671 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2.1101 | 0 | 0 | 0 | 0 | 0 | 0.5737 | 0 |
| ?2 | 3.7613 | 7.3281 | 0 | 5.3598 | 2.8093 | 6.4777 | 13.9372 | 3.5684 | 40.3762 | 9.0914 | 9.0853 | 4.7409 | 3.974 | 2.3904 | 7.0211 | 11.2489 | 0 | 19.5722 | 6.3454 | 6.4641 |
| ?3 | 2.8959 | 5.5218 | 0.6636 | 0 | 4.545 | 2.8922 | 6.8144 | 5.3975 | 41.0665 | 3.6238 | 6.0806 | 12.1334 | 8.6865 | 7.8033 | 5.6677 | 17.3953 | 5.1887 | 17.4112 | 18.3247 | 13.092 |
| ?4 | 117.7889 | 443.1501 | 144.1446 | 92.6276 | 136.3486 | 238.557 | 91.604 | 211.2791 | 100.4809 | 27.5497 | 101.9194 | 105.918 | 78.537 | 120.6983 | 212.0846 | 70.1299 | 106.1415 | 150.7324 | 103.2415 | 149.5243 |
| ?5 | 1754.636 | 1546.397 | 4391.827 | 868.8195 | 685.4201 | 1752.167 | 1229.95 | 1456.665 | 966.0655 | 1691.126 | 1220.853 | 1279.229 | 2167.048 | 1399.592 | 1939.577 | 978.7596 | 1754.717 | 1110.225 | 1262.765 | 1477.273 |
| ?6 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ?7 | 192.1065 | 306.1955 | 15.9268 | 66.9972 | 157.3819 | 32.1699 | 73.4717 | 49.6115 | 426.9924 | 745.9603 | 48.8152 | 402.5713 | 229.9982 | 266.8196 | 120.8459 | 472.8729 | 135.8491 | 640.3191 | 553.0694 | 511.0994 |
| ?8 | 0 | 2.9371 | 0 | 0 | 0.7354 | 0 | 0 | 0.5977 | 1.1635 | 1.0596 | 0 | 0.8035 | 0 | 0.5097 | 0.6042 | 0.4855 | 0 | 1.4503 | 4.5898 | 0.5285 |
| ?9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
3. 绘制箱型图
ndata.iloc[1000:1010, ].transpose().plot(kind='box', title='Normal Sample Gene Boxplot', rot=30)
plt.tick_params(labelsize=10)
plt.savefig('Normal_Sample_Gene_box_plot', bbox_inches='tight')
plt.show()绘制正常样本中部分基因的箱型图,并保存为图片文件。

4. 删除表达量低于阈值的基因
ndata = ndata.iloc[29:, :]
tdata = tdata.iloc[29:, :]删除正常样本和肿瘤样本中表达量低于阈值的基因。
5. 计算差异显著的基因
gene_sig = ndata[ndata.mean(axis=1) > num_cutoff].index.intersection(tdata[tdata.mean(axis=1) > num_cutoff].index)
Ndata = ndata.loc[gene_sig]
Tdata = tdata.loc[gene_sig]选择正常样本和肿瘤样本中表达量高于阈值的基因,并保存为新的数据。
6. 初始化结果数据表格
p_value = [1.] * len(Ndata)
log2FoldChange = []
label = ['0'] * len(Ndata)
result = pd.DataFrame([p_value, log2FoldChange, label])
result = result.transpose()
result.columns = ['p_value', 'log2FC', 'label']
result.index = Ndata.index.tolist()
p = []创建一个结果数据表格,包含 p 值、log2 fold change 和标签,并初始化为默认值。
7. 进行秩和检验和差异倍数计算
for i in Ndata.index:
p1 = ranksums(Tdata.loc[i], Ndata.loc[i], alternative='greater')[1]
p2 = ranksums(Tdata.loc[i], Ndata.loc[i], alternative='less')[1]
p3 = ranksums(Tdata.loc[i], Ndata.loc[i])[1]
result.loc[i, 'log2FC'] = np.log2(np.average(Tdata.loc[i]) / np.average(Ndata.loc[i]))
p.append(p3)
if (p1 <= p_cutoff):
result.loc[i, 'p_value'] = p1
if result.loc[i, 'log2FC'] > np.log2(FC_cutoff):
result.loc[i, 'label'] = '1'
if (p2 <= p_cutoff):
result.loc[i, 'p_value'] = p2
if result.loc[i, 'log2FC'] < -np.log2(FC_cutoff):
result.loc[i, 'label'] = '-1'对每个基因进行秩和检验,计算 p 值和差异倍数,并根据设定的阈值确定差异显著的基因,并给予标签。
8. 可视化分析
print('finished')
plt.hist(result['log2FC'], bins=10, color='blue', alpha=0.6, edgecolor='black')
plt.title("fold change")
plt.show()

9. 代码整合
import numpy as np
import pandas as pd
import scipy
import matplotlib.pyplot as plt
import seaborn as sns
from scipy.stats import zscore
from scipy.stats import ranksums
from pyHSICLasso import HSICLasso
from sklearn.preprocessing import LabelEncoder
p_cutoff = 0.001
FC_cutoff = 3
num_cutoff = 10
# 读取数据ndata normal data,tdata tumor data
ndata = pd.read_csv("normal_data.csv", index_col=0, header=0)
tdata = pd.read_csv("tumor_data.csv", index_col=0, header=0)
# 箱型图
# 中位数
# QL称为下四分位数,表示全部观察值中有四分之一的数据取值比它小;
# QU称为上四分位数,表示全部观察值中有四分之一的数据取值比它大;
# IQR称为四分位数间距,是上四分位数QU与下四分位数QL之差,其间包含了全部观察值的一半。
# 异常值通常被定义为小于QL-1.5IQR或大于QU+1.5IQR的值。
ndata.iloc[1000:1010, ].transpose().plot(kind='box', title='Normal Sample Gene Boxplot', rot=30)
plt.tick_params(labelsize=10)
plt.savefig('Normal_Sample_Gene_box_plot', bbox_inches='tight')
plt.show()
# delete gene expression < num
ndata = ndata.iloc[29:, :]
tdata = tdata.iloc[29:, :]
gene_sig = ndata[ndata.mean(axis=1) > num_cutoff].index.intersection(tdata[tdata.mean(axis=1) > num_cutoff].index)
Ndata = ndata.loc[gene_sig]
Tdata = tdata.loc[gene_sig]
p_value = [1.] * len(Ndata)
# log2 fold change的意思是取log2,这样可以可以让差异特别大的和差异比较小的数值缩小之间的差距
log2FoldChange = []
label = ['0'] * len(Ndata)
result = pd.DataFrame([p_value, log2FoldChange, label])
result = result.transpose()
result.columns = ['p_value', 'log2FC', 'label']
result.index = Ndata.index.tolist()
p = []
# 秩和检验(也叫Mann-Whitney U-test),检验两组数据是否来自具有相同中位数的连续分布,检验它们是否有显著差异。
# ’less‘ 基于 x 的分布小于基于 y 的分布
# ‘greater’ x 的分布大于 y 的分布。
for i in Ndata.index:
p1 = ranksums(Tdata.loc[i], Ndata.loc[i], alternative='greater')[1]
p2 = ranksums(Tdata.loc[i], Ndata.loc[i], alternative='less')[1]
p3 = ranksums(Tdata.loc[i], Ndata.loc[i])[1]
result.loc[i, 'log2FC'] = np.log2(np.average(Tdata.loc[i]) / np.average(Ndata.loc[i]))
p.append(p3)
if (p1 <= p_cutoff):
result.loc[i, 'p_value'] = p1
if result.loc[i, 'log2FC'] > np.log2(FC_cutoff):
result.loc[i, 'label'] = '1'
if (p2 <= p_cutoff):
result.loc[i, 'p_value'] = p2
if result.loc[i, 'log2FC'] < -np.log2(FC_cutoff):
result.loc[i, 'label'] = '-1'
print('finished')
# 查看差异倍数fold分布
plt.hist(result['log2FC'], bins=10, color='blue', alpha=0.6, edgecolor='black')
plt.title("fold change")
plt.show()
# result.to_csv('result.csv')
up_data_n = Ndata.loc[result.query("(label == '1')").index]
up_data_t = Tdata.loc[result.query("(label == '1')").index]
down_data_n = Ndata.loc[result.query("(label == '-1')").index]
down_data_t = Tdata.loc[result.query("(label == '-1')").index]
data = pd.concat([pd.concat([up_data_n, up_data_t], axis=1), pd.concat([down_data_n, down_data_t], axis=1)], axis=0)
deg_gene = pd.DataFrame(data.index.tolist())
deg_gene.to_csv('deg_gene.csv')
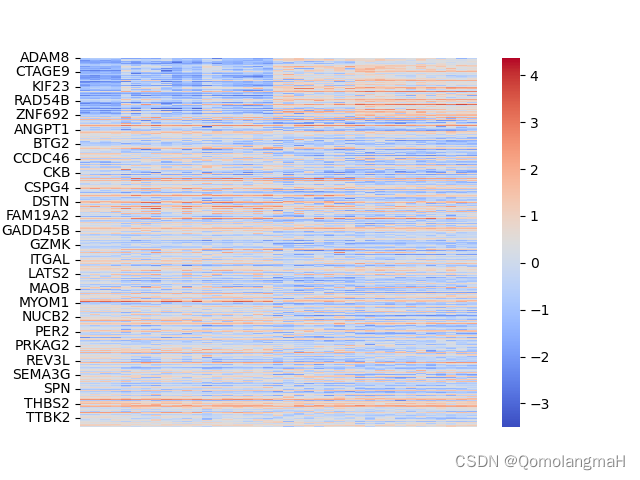
z1 = zscore(np.log2(data+1), axis=0)
sns.heatmap(data=z1, cmap="coolwarm", xticklabels=False)
plt.savefig('heatmap_plot', bbox_inches='tight')
plt.show()
p = -np.log10(p)
ax = sns.scatterplot(x="log2FC", y=p,
hue='label',
hue_order=('-1', '0', '1'),
palette=("#377EB8","grey","#E41A1C"),
data=result)
ax.set_ylabel('-log(pvalue)', fontweight='bold')
ax.set_xlabel('FoldChange', fontweight='bold')
plt.savefig('deg_p_fc_plot', bbox_inches='tight')
plt.show()