8 分子相互作用的人工智能
正如在第 5、6 和 7 节中所描述的那样,人工智能已经彻底改变了分子学习、蛋白质科学和材料科学领域。尽管已经广泛研究了用于单个分子的人工智能,但分子的物理和生物功能通常是由它们与其他分子的相互作用驱动的。在本节中,我们进一步介绍了用于分子相互作用的人工智能,特别关注小分子与蛋白质或材料之间的相互作用。
8.1 概述
作者:Meng Liu,Shuiwang Ji
这个研究领域侧重于使用人工智能来深入了解小分子与其他物质之间相互作用的分子机制,这对于推进我们对分子相互作用的理解并为生命科学和材料科学中的一系列挑战提供实际解决方案具有巨大潜力。
对于分子-蛋白质和分子-材料相互作用,如图 30 所示,我们将现有任务分为预测任务和生成任务两类。请注意,我们的分类是基于任务的性质而不是执行任务的方法。具体来说,与小分子和蛋白质的相互作用相关的预测任务是结合(或对接)预测。结合是指一种小分子,即配体,基于它们的本地形状互补和化学相互作用[费希尔 1894],也被称为“锁-钥”模型,与目标蛋白质结合的过程。目标蛋白质通常与人类疾病相关联。药物分子结合到目标蛋白质上,以抑制或激活它以治疗人类疾病。这个任务包括结合位姿预测和结合亲和力预测。另一方面,对于这种相互作用的生成任务是生成可以与给定目标蛋白质结合的分子,称为基于结构的药物设计[Anderson 2003]。蛋白质-配体结合预测和基于结构的药物设计都是药物发现中的基本且具有挑战性的问题。就小分子和材料的相互作用而言,据我们所知,现有工作中只研究了预测任务。具体而言,我们感兴趣的是从分子-材料对结构中预测吸附能(S2E)和每个原子的力(S2F)。此外,鉴于分子-材料对的初始结构,迫切需要预测其松弛最终结构(IS2RS)和在其松弛状态下的吸附能(IS2RE)。这些任务对于材料科学中的许多问题至关重要,例如可再生能源存储的电催化剂设计[Zitnick等人 2020]。分子-材料相互作用的生成任务仍未开发,我们将在第 8.4.5 节讨论潜在机会。本节中涵盖的方法概述如图 31 所示。
确保在三维空间中保持可取的对称性是分子相互作用任务的重要方面。这种独特的对称性与考虑单个分子不同,因为它包括涉及相互作用的多个实例。特别是,在整个相互作用系统的背景下,考虑每个分子的对称性属性是至关重要的。
8.2 蛋白质-配体结合预测
作者:Hannes Stärk,Yuchao Lin,Shuiwang Ji,Regina Barzilay,Tommi Jaakkola
推荐先修课程:第 5.2 节,第 6.3 节
在本节中,我们研究蛋白质-配体结合问题。我们的目标是推断小分子,可能是药物,与蛋白质如何相互作用。为此,我们讨论分子对接和结合亲和力预测,这两者对于药物发现或分子生物学等领域都非常重要。
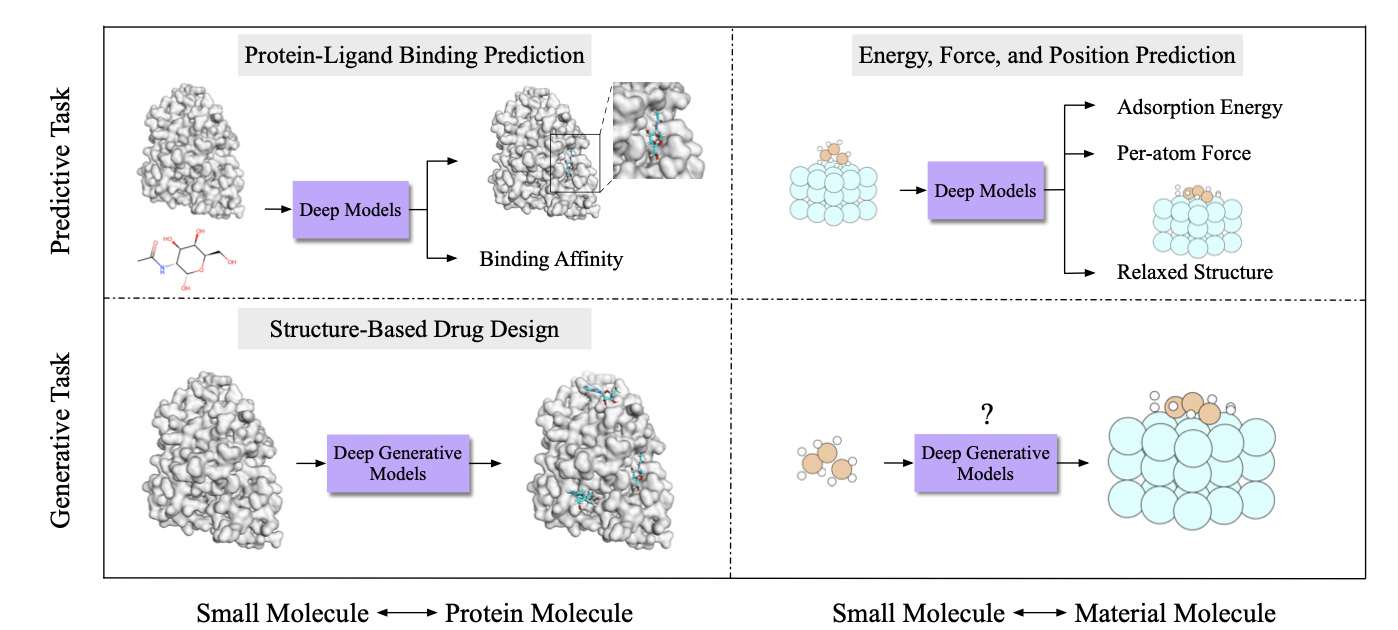
图30. 分子相互作用中我们涵盖的任务示意图。双向箭头表示相互作用。对于分子-蛋白质相互作用和分子-材料相互作用,我们将现有任务分为预测任务和生成任务两类。在蛋白质-配体结合预测中,我们旨在预测配体的结合位姿和结合强度,即结合亲和力。在基于结构的药物设计中,我们希望生成可以与给定目标蛋白质结合的3D配体分子。在分子-材料对的预测任务中,我们感兴趣的是在给定分子-材料对结构的情况下预测吸附能和每个原子的力矢量。此外,我们也关注在输入初始结构的情况下预测松弛最终结构。分子-材料相互作用的生成任务尚未开发,我们将在第8.4.5节讨论可能的方向。
8.2.1 问题设置
在对接中,我们会获得蛋白质结构(其氨基酸标识和原子坐标)以及小分子(配体)的分子图。目标是预测配体最有可能与蛋白质结合的原子位置。这个任务可以分为两种情况,一种是对接位置(口袋)大致已知的情况,另一种是没有任何先前知识的盲目对接情况。在任何一种情况下表现良好都意味着有高比例的大致正确的预测。同时,对于结合强度的预测,我们指的是预测一个排名或标量的结合亲和力,它表示配体与蛋白质结合的强度;即,配体和蛋白质在结合状态与非结合状态下出现的比例。用于此任务的输入可以是结合的蛋白质-配体结构或蛋白质结构和配体的分子图。
在对接和结合强度预测中,考虑问题设置的讨论细节非常重要。此外,在这两个任务中,重要的是在绑定到配体时是否可以访问蛋白质的结构(全构象,holo-structure),未结合的蛋白质结构(无配体构象,apo-structure),与另一个配体结合的蛋白质的结构,或者仅从AlphaFold2等计算生成的结构。在对接评估中,通常假定已知结合的结构,这在应用中是不切实际的,因此比较其他情况是可取的。
为了在正式的上下文中介绍对接和结合强度预测,配体的结构可以表示为 M = (𝐴,𝐸,𝐶),其中 𝐴 = [𝒂1,··· ,𝒂𝑛] 表示所有分子中包含的 𝑛 个原子的原子属性,例如原子类型。边缘特征,可能包括键的类型和键长,用 𝐸 = [𝒆1, · · · , 𝒆𝑙 ] 表示分子中所有 𝑙 个化学键。与此同时,3D 坐标矩阵表示为 𝐶 = [𝒄1, · · · , 𝒄𝑛 ] ∈ R3×𝑛 。
类似地,蛋白质或已知口袋的结构可以表示为 P = (𝐵,𝑆)。这里,𝐵 = [𝒃1,··· ,𝒃𝑚] 表示指定蛋白质或口袋中的所有氨基酸或原子的节点特征,具体取决于详细说明的粒度级别,如第 6 节所述。另外,与蛋白质中的氨基酸的α-碳或结构中的原子相对应的3D坐标由 𝑆 = [𝒔1, · · · , 𝒔𝑚 ] ∈ R3×𝑚 表示。
在对接中,主要目标是基于蛋白质/口袋结构从对接姿态的分布中进行采样,可以表示为 𝑝pose(𝐶|𝐵,𝑆,𝐴,𝐸)。方法可以直接建模分布 𝑝pose(𝐶|𝐵,𝑆,𝐴,𝐸) 然后进行采样,或者预测配体构象的𝑘个几何结构,使得 𝑓 pose (𝐵, 𝑆, 𝐴, 𝐸) ↦→ [𝐶1, · · · , 𝐶𝑘 ]。
另一方面,结合强度预测的目标是估计结合亲和力,提供排名或进行二进制的结合 vs. 非结合预测。我们将这些目标统一表示为 𝑞,其中 𝑞 ∈ R 或 𝑞 ∈ Z+ 或 𝑞 ∈ [0, 1],任务表示为 𝑓strength(𝐴,𝐸,𝐶,𝐵,𝑆) ↦→ 𝑞。需要注意的是,在结合强度预测中,可能不提供配体或蛋白质/口袋的几何信息。
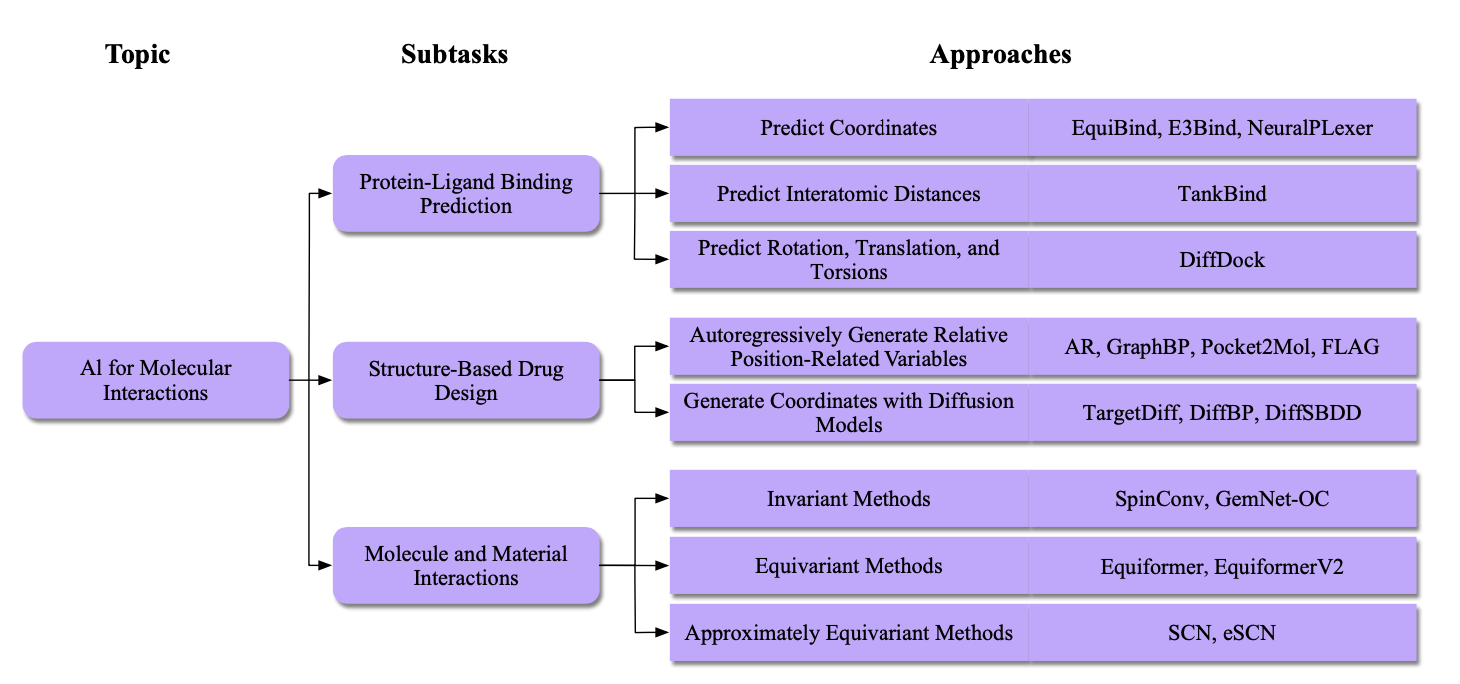
图31. 分子相互作用中的任务和方法概述。本节考虑三个任务,包括蛋白质-配体结合预测、基于结构的药物设计以及分子-材料对的能量、力和位置预测。在蛋白质-配体结合预测中,一类方法,包括EquiBind [Stärk等人 2022b]、E3Bind [Zhang等人 2023a] 和NeuralPLexer [Qiao等人 2023],旨在直接预测配体的3D坐标。相比之下,TankBind [Lu等人 2022] 预测蛋白质片段与配体之间的原子间距。此外,DiffDock [Corso等人 2022] 生成由RDKit提供的种子构象的旋转、平移和扭转角度。在基于结构的药物设计中,一类方法,包括AR [Luo等人 2021]、GraphBP [Liu等人 2022c]、Pocket2Mol [Peng等人 2022] 和FLAG [Zhang等人 2023b],旨在通过建模它们的相对位置相关变量来自回归地生成配体原子/片段。另一类方法,包括TargetDiff [Guan等人 2023]、DiffBP [Lin等人 2022b] 和DiffSBDD [Schneuing等人 2022],考虑通过扩散模型直接生成所有配体原子的3D坐标。对于分子-材料对的预测任务,不变方法包括SpinConv [Shuaibi等人 2021] 和GemNet-OC [Gasteiger等人 2022],而等变方法包括Equiformer [Liao和Smidt 2023] 和EquiformerV2 [Liao等人 2023]。此外,SCN [Zitnick等人 2022] 和eSCN [Passaro和Zitnick 2023] 是近似等变方法。
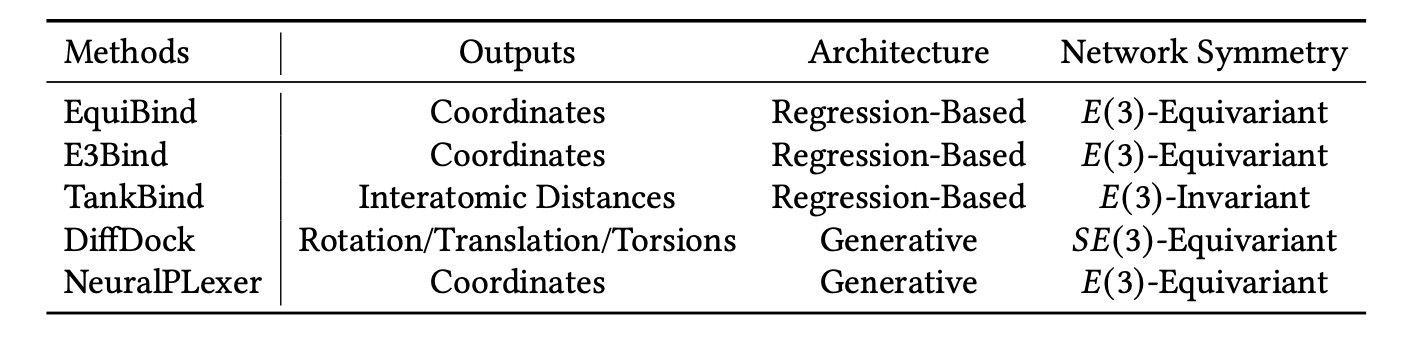
表29. 盲目蛋白质-配体对接的深度学习方法概述(涉及对称性)。EquiBind [Stärk等人 2022b] 和E3Bind [Zhang等人 2023a] 应用 𝐸(3)-等变网络来预测配体坐标。TankBind [Lu等人 2022] 估计蛋白质口袋候选区域与配体之间的原子间距,并为每个候选区域估计亲和力。DiffDock [Corso等人 2022] 利用 𝑆𝐸(3)-等变扩散模型,涵盖旋转、平移和扭转,以在对候选区域进行排序之前对其进行采样。NeuralPLexer [Qiao等人 2023] 预测接触图并将其应用于 𝐸(3)-等变扩散模型,以生成配体坐标。
8.2.2 技术挑战
在我们描述分子对接时,我们努力预测配体的最可能的结合位姿。这仅仅是描述每个可能的配体构象在蛋白质条件下的概率的玻尔兹曼分布的全局模式。理想情况下,我们希望有一个生成模型,能够复制这个高维分布,但具有稀疏的支持,考虑到通常只有一个模式的数据。在给定可信任的配体构型的大空间的情况下,生成最低能量(即概率最高)的构象已经是一个困难的问题。
与蛋白质结构预测相比,对于对接来说,一个挑战是,对接方法无法依赖于大量的序列数据来获取进化信息以限制可能结构的集合,这在几何深度学习对分子对接产生重大影响之前,部分解释了蛋白质结构预测的成功。数据方面也存在另一个技术挑战;具有合理质量的易于获取的训练数据量仅为20k个样本。虽然PDB中有更多的复合物,但要从中清除专家知识和难以处理的数据,例如仅具有非常低亲和力的虚假相互作用的复合物,这是困难的。
类似的数据问题也阻碍了对结合强度的预测进展,因为对于3D结构,只有很少的数据点(约20k个)和嘈杂的测量(也包括基于序列的数据),阻止了成功的亲和力预测器适用于一般的蛋白质-配体组合。然而,一个已经在当前有用的策略是训练针对特定蛋白质的预测器,只需将配体作为输入(在具体蛋白质的情况下,前提是可以获得足够的结合亲和力数据,或者可以在主动学习设置中收集数据)。通向一般蛋白质-配体亲和力预测的潜在方向可能是使用几何深度学习来辅助或近似于用于计算结合自由能的统计力学方法。但这再次受到建模玻尔兹曼分布及其分区的困难性的制约。
最后,我们讨论了这两个任务中涉及的对称性。对接是一个 𝑆𝐸(3)-等变任务;旋转或平移输入蛋白质结构应该导致生成的配体姿态相应地进行旋转和平移。从技术上讲,对于旋转平移 𝑔 ∈ 𝑆𝐸(3) 及其对应的群作用▷,这个任务要求 𝑝pose(𝑔▷𝐶|𝐵,𝑔▷𝑆,𝐴,𝐸) = 𝑝pose(𝐶|𝐵,𝑆,𝐴,𝐸) 和 𝑓 pose (𝐵, 𝑔▷𝑆, 𝐴, 𝐸) = 𝑔▷ 𝑓 pose (𝐵, 𝑆, 𝐴, 𝐸)。相反,结合强度预测是一个 𝑆𝐸 (3)-不变任务,因为对系统的旋转和平移不会影响预测。从形式上讲,对于旋转平移 𝑔 ∈ 𝑆𝐸(3),希望 𝑓strength(𝐴,𝐸,𝑔▷𝐶,𝐵,𝑔▷𝑆) = 𝑓strength(𝐴,𝐸,𝐶,𝐵,𝑆)。
8.2.3 现有方法。
我们将蛋白质-配体对接模型分为三种不同类型:传统的基于搜索的对接、基于回归的对接和生成对接。为了突出深度学习在盲目蛋白质-配体对接领域取得的最新进展,我们提供了一个关于它们对对称性处理的盲目蛋白质-配体对接方法概述,具体见表格29。
传统基于搜索的对接:传统方法采用𝐸(3)不变的评分函数,将可能的配体姿势分配概率,并使用优化算法来找到评分函数的全局最小值,即最可能的姿势[Trott和Olson 2010;Koes等人 2013;Halgren等人 2004]。最常见的评分函数包括基于物理的不变量的项,如原子间距离,并使用很少的学习参数。最近,也出现了一些深度学习参数化方法,例如使用了3D卷积神经网络[McNutt等人 2021]。重要的是,这些方法大多是为已知口袋进行对接开发的(有例外[Hassan等人 2017]),在盲目对接中面临更大的搜索空间,导致推断时间较长。此外,它们的评分函数对于输入蛋白质与结合结构的偏差敏感[Corso等人 2022;Karelina等人 2023],这限制了它们对于与计算生成或未结合的蛋白质结构进行对接的能力。
基于回归的对接:更近期的深度学习方法通过直接使用𝐸(3)等变/不变的图神经网络来预测配体结合姿势,显著加快了盲目对接的速度,而不是为搜索算法参数化评分函数。在基于回归的方法中,EquiBind [Stärk等人 2022b] 通过找到表征结合口袋的关键点并将配体与这些关键点叠加来产生预测。与此同时,Tankbind [Lu等人 2022] 将蛋白质分成口袋候选项,并预测每个候选项的蛋白质-配体距离和亲和力得分。E3Bind [Zhang等人 2023a] 使用相同的口袋候选项,但为它们生成了成对表示,然后通过更新初始配体坐标来迭代解码这些表示。这些基于回归的方法的一个缺点是,它们被迫预测单一的姿势,尽管可能有多个配置是合理的,在玻尔兹曼分布下具有显著的可能性。这通常会导致预测出现不合理的情况,如立体位阻和自相交[Corso等人 2022]。
生成对接:为了解决对接任务和基于回归的解决方案之间的不匹配,首次提出了DiffDock [Corso等人 2022] 作为生成模型。其扩散模型由Tensor Field Networks [Thomas等人 2018] 参数化,并在生成的样本上使用置信度模型进行排名。与此同时,NeuralPLexer [Qiao等人 2023] 预测了一个接触图,条件是𝐸(3)等变扩散模型生成了配体坐标,并从未结合的结构重新折叠蛋白质结构到结合的结构。提到的生成模型也能够以合理的准确性对接到未结合或计算生成的蛋白质结构。
8.2.4 数据集和基准。
一个重要的小分子与蛋白质结合的3D结构数据集是PDBBind [Liu等人 2017],它从蛋白质数据银行(PDB)[Berman等人 2003]中筛选出具有结合亲和力并符合其他质量标准的复合物。该数据集包含20,000个复合物,涵盖了4,000个独特的蛋白质。一个常见的数据集分割[Stärk等人 2022b] 是基于时间的,将2019年之前的复合物用作训练数据,将更新的复合物用作测试数据。与PDBBind的选择标准相比,BindingMOAD [Wagle等人 2023] 应用了更少的严格标准,从PDB中提取了40,000个蛋白质-配体结构。APObind [Aggarwal等人 2021] 为其蛋白质-配体复合物提供了未结合的蛋白质结构。对于肽类的方法,Propedia [Martins等人 2021] 从PDB中提取了蛋白质-肽复合物。虽然结构数据的数量有限,但在ChEMBL [Mendez等人 2019] 中有更多的蛋白质-配体结合亲和力数据,共有2000万个活性测量数据。
为了评估对接预测,通常会估计正确预测的比例,这被定义为生成的配体姿势与真实结构的RMSDT在指定阈值以下的情况。此外,生成结构中的立体位阻数量是
一个相关的度量标准。为了衡量结合强度预测的性能,度量标准取决于任务,如二元分类的准确性(结合与不结合)、正确排名一组配体结合强度的排名相关性,或结合亲和力的均方误差(MAE)预测。为了更好地模拟真实世界的对接问题,该领域希望评估对未结合或计算生成的蛋白质结构(apo-结构)的对接,而不是假定输入为结合结构。
8.2.5 开放研究方向。
尽管分子对接领域取得了令人印象深刻的进展,但这个任务仍然远未解决,例如,在对接到来自ESMFold的结构时,DiffDock的准确性仅为22%。可能的改进可以源于更好的生物物理结构生成模型、更有意义的特征嵌入,或更具表现力的3D体系结构。然而,结构预测能力为将其与下游结合强度预测器集成提供了一条道路。解锁这些方法或类似方法,共同利用可用的结构数据和更大量的基于序列的结合亲和力数据是有希望的,初步的成功案例已经存在[Moon等人 2023]。进一步帮助访问这些更大量的数据的方法可以是创造性的、问题特定的方法,用于处理亲和力测量中的噪音。
此外,将分子对接扩展到模拟蛋白质在结合过程中的构象变化具有很大的价值。我们认为这更有意义和现实,因为实际上结合通常会改变蛋白质的构象。最后,我们希望引起人们对生成模型在统计力学方法中计算或比较蛋白质-配体相互作用强度/概率的潜力的关注,而不是依赖于实验亲和力测量的回归。
8.3 基于结构的药物设计
作者:刘蒙,傅天凡,迈克尔·布朗斯坦,孙济萍,水旺基 推荐先修知识:第5.2节,第6.3节
在这一部分中,我们考虑基于结构的药物设计(SBDD),这是一项用于蛋白质-配体相互作用的生成任务。在这个任务中,我们的目标是生成3D分子,即配体,它们可以紧密结合到特定的蛋白质(也称为目标蛋白质),这可以被制定为一个条件生成问题。
8.3.1 问题设置。
正式地,根据第8.2节,我们让M = (𝐴,𝐸,𝐶)表示配体分子,P = (𝐵,𝑆)表示蛋白质结合位点。总体上,这个任务的目标是从观察到的蛋白质-配体对中学习条件分布 𝑝(M|P)。
8.3.2 技术挑战。
独特的对称性挑战是由于这个生成任务涉及多个分子相互作用,而不是单个分子,所以相对于建模单个分子,这导致了更复杂的对称性挑战。特别地,必须考虑单个分子的对称性,以及它们相对于彼此的相对位置和方向。具体来说,如果我们旋转或平移蛋白质结合位点,生成模型生成的分子应相应地旋转或平移。从数学上讲,学习的条件分布应满足 𝑝(M|P) = 𝑝(𝑔 ▷ M|𝑔 ▷ P),其中 𝑔 ∈ 𝑆𝐸(3),▷表示其相应的群作用。为了实现这一点,模型生成的分子应对蛋白质的𝑆𝐸(3)变换具有等变性。
此外,这个任务面临着极其广阔的搜索空间的挑战。具体来说,包含所有可能分子的化学空间估计超过1060。此外,3D分子还有额外的构象空间。然而,只有极小部分空间与药物发现相关,因为分子需要满足特定的标准才能被认为是“药物样”。因此,在考虑与目标蛋白质的相互作用时,如何有效地建模和探索这样的空间是这个任务中的一个基本考虑因素。
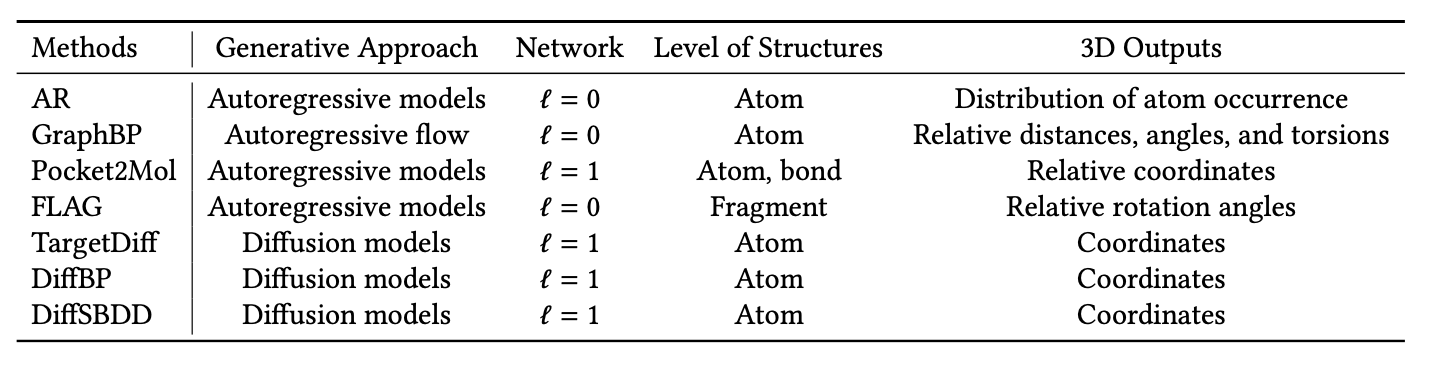
表格30. 基于结构的药物设计现有方法总结,涵盖采用的生成方法、使用的网络、建模结构的级别和3D输出变量。在这些方法中,AR [Luo等人 2021]、GraphBP [Liu等人 2022c] 和Pocket2Mol [Peng等人 2022] 通过建模相对位置相关的变量,自回归地生成原子,这些变量可以用来确定新原子的位置。另外,FLAG [Zhang等人 2023b] 不是生成原子,而是考虑自回归地生成片段。相比之下,TargetDiff [Guan等人 2023]、DiffBP [Lin等人 2022b] 和DiffSBDD [Schneuing等人 2022] 使用扩散模型直接一次性生成所有原子的3D坐标。
8.3.3 现有方法。
早期的研究要么在目标蛋白质的3D信息的条件下生成分子SMILES字符串[Skalic等人 2019;Xu等人 2021c],要么使用估计的对接分数作为奖励函数来引导分子生成模型[Li等人 2021d;Fu等人 2022a]。它们没有明确地在3D空间中建模配体分子与目标蛋白质之间的关键相互作用。Ragoza等人 [2022] 将蛋白质-配体复合物结构转化为原子密度格点,然后使用生成方法处理3D图像数据来解决这个任务。一个限制是没有保留前述的等变性属性,因为3D卷积神经网络[Ji等人 2013]对于3D格点数据不是𝑆𝐸(3)等变操作。
最近,随着几何深度学习和生成建模的发展,蛋白质-配体复合物被自然地建模为3D几何体,它们复杂的相互作用和对称性约束可以被有效地编码。一般来说,我们可以将这些最新的方法分为两类。第一类方法基于当前上下文生成原子自回归地,包括结合位点和先前生成的原子。为了保持前述的所需𝑆𝐸(3)等变性质,这些方法在每个自回归步骤中考虑相对于当前上下文的新原子的相对位置相关变量的建模,而不是直接生成其3D坐标。具体来说,AR [Luo等人 2021] 使用不变的3D图神经网络(特征阶数𝑙 = 0)来建模原子在3D位置上出现的分布,将查询位置与当前上下文的相对距离作为输入。使用与上下文相关的不变距离和不变的3D图神经网络确保了建模的分布对于上下文的旋转和平移是等变的。相比之下,GraphBP [Liu等人 2022c] 和Pocket2Mol [Peng等人 2022] 在每一步中都模拟新原子相对于所选焦点原子的相对位置。具体来说,GraphBP首先在焦点原子处构建一个局部球面坐标系(SCS),它对于上下文的旋转和平移是等变的。然后,通过不变的3D图神经网络生成相对于参考SCS的不变距离、角度和扭转。Pocket2Mol使用一个等变的神经网络(特征阶数𝑙 = 1)作为编码器,焦点原子的等变特征可以用来等变地生成新原子的相对位置。Pocket2Mol还明确生成键。FLAG [Zhang等人 2023b] 不使用原子作为构建块,而是考虑分段生成3D分子。这种分段词汇可以从化学先验中获得,并有助于生成有效且逼真的分子。在每一步中,FLAG将新片段组装到当前上下文中,然后预测新片段相对于所选焦点片段的旋转角度。与GraphBP类似,FLAG可以保持𝑆𝐸(3)等变性。在上述方法中,AR、Pocket2Mol和FLAG通过掩码填充模式进行训练,其中原子或片段被随机掩码,模型被训练以恢复它们。相比之下,GraphBP通过最大化原子放置步骤轨迹的对数似然来进行训练,这得益于流模型的精确似然计算。
另一类方法,如TargetDiff [Guan等人 2023]、DiffBP [Lin等人 2022b] 和DiffSBDD [Schneuing等人 2022],考虑直接生成所有原子的3D坐标。与上述自回归采样方法相比,这种一次性生成方式不需要原子之间的顺序,并且可以考虑整个配体分子的全局相互作用。在EDM [Hoogeboom等人 2022]的框架下,这些方法在连续和离散空间中应用扩散模型[Ho等人 2020]来建模原子坐标和原子类型。去噪步骤由一个等变的GNN(特征阶数𝑙 = 1)[Satorras等人 2021b]来建模。为了绕过在扩散过程中维持平移等变性的困难,它们将系统的质心(CoM)平移到仅在线性子空间中扩散和去噪坐标。此外,采用宽松的表示法,由于潜变量遵循旋转不变的高斯分布𝑝(𝒓𝑇 ),并且过渡分布𝑝(𝒓𝑡−1|𝒓𝑡, P)是等变的,前述的等变性质可以得到满足[Köhler等人 2020]。
除了深度生成模型,强化学习(RL)也被用于基于结构的药物设计[Li等人 2021d;Fu等人 2022a],它将药物分子生成过程形式化为马尔可夫决策过程(MDP)。与显式构建连续数据分布的生成模型不同,强化学习从离散空间中选择动作,该动作将获得最大的奖励(例如,结构基于药物设计中的对接分数)。具体来说,DeepLigBuilder [Li等人 2021d] 构建了一个策略网络,以选择适当的动作(是否/在何处添加原子,添加哪个原子)来在目标位点中生长分子;强化遗传算法(RGA) [Fu等人 2022a] 利用策略网络智能地选择遗传算法(GA)的离散动作空间(突变,交叉位置),从而抑制了遗传算法中的随机漫步行为。
值得一提的是,早期基于SMILES、SELF-IES或分子图的分子优化方法,如果将对接分数作为优化目标[Huang等人 2021;Gao等人 2022],也可以在基于结构的药物设计中取得竞争性的性能,包括SMILES变分自动编码器(SMILES-VAE)[Gómez-Bombarelli等人 2018]、接合树变分自动编码器(JTVAE)[Jin等人 2018]、图卷积策略网络(GCPN)[You等人 2018b]、分子图级遗传算法(Graph-GA)[Jensen 2019]、图自回归流(GraphAF)[Shi等人 2019]、多约束分子采样(MIMOSA)[Fu等人 2021]等方法。
8.3.4 数据集和基准。
首先,我们简要介绍几个基于结构的药物设计数据集,包括CrossDocked2020、PDBBing、DUD-E和scPDB。
(1) CrossDocked2020 [Francoeur等人 2020] 是一个广泛使用的基于结构的药物设计方法性能评估基准数据集。CrossDocked2020包含了22,584,102个对接的蛋白质-配体复合物。这些复合物是通过交叉对接生成的,其中使用Pocketome [Kufareva等人 2012]将与特定口袋相关联的配体通过smina对接软件 [Koes等人 2013]对接到由该口袋分配的每个受体中。CrossDocked2020总共包括2,922个口袋和13,839个配体。鉴于这些复合物的质量变化,现有研究中通常包括一个筛选步骤。这一步涉及删除结合位姿的均方根偏差(RMSD)超过一定阈值的复合物。这旨在鼓励模型生成具有更高结合亲和力的配体分子。
(2) PDBBind是从蛋白质数据银行(PDB)[Berman等人 2003]衍生出的大型存储库,包含了蛋白质-配体复合物的实验确定的结合亲和力数据[Wang等人 2004]。它包括19,445个蛋白质-配体对。
(3) 有用的假象目录,增强版(DUD-E)提供了一个用于蛋白质-配体对接的有用假象目录[Mysinger等人 2012]。它包含22,886个蛋白质-配体复合物和它们对102个不同蛋白质靶点的亲和力。
(4) scPDB是专门为基于结构的药物设计定制的蛋白质数据银行(PDB)的精炼版本,可用于识别蛋白质-配体对接的最佳结合位点[Meslamani等人 2011]。它包含16,034个蛋白质-配体对,涉及4,782个蛋白质和6,326个配体。
为了评估不同生成方法的性能,通常使用几类指标。第一类涉及测量生成分子的质量,包括它们的化学有效性、新颖性和多样性。此外,比较生成分子与参考集之间特定变量的分布,如键长[Ragoza等人 2022;Liu等人 2022c;Peng等人 2022]、键角[Ragoza等人 2022]和不同模式的出现[Peng等人 2022],可以提供有关生成分子质量的进一步见解。第二类旨在使用Vina能量或基于深度学习的评分函数[Ragoza等人 2022]来估计生成分子与目标蛋白质之间的结合亲和力。将生成分子的结合亲和力与参考分子的结合亲和力进行比较有助于评估它们在与目标结合方面的有效性。最后一类包括测量其他重要性质,如药物样性QED(药物样性的定量估计)[Bickerton等人 2012]和SA(合成可及性)[Ertl和Schuffenhauer 2009]。[Huang等人 2021]包括了一个基于结构的药物设计基准,比较了五种机器学习方法在相同数量的对接预言器调用(5K)下的性能。
8.3.5 开放性研究方向。
尽管深度学习方法在基于结构的药物设计中取得了进展,但如何有效和高效地建模庞大的化学空间以生成有效和可合成的分子仍然是一个主要挑战。包括基本的化学先验,如基本片段和支架,可能是解决这一挑战的一个方向。例如,为了实现基于片段的药物设计,DiffLinker [Igashov等人 2022] 使用了一个𝐸(3)-等变的3D条件扩散模型,类似于DiffSBDD,将不相连的分子片段(药效团)链接成单一分子,同时可以考虑周围的蛋白质口袋作为条件信息。此外,最近的一项工
作[Adams和Coley 2022]提出了一种基于形状的3D分子生成方法,这可能是缩小建模空间的另一个有前途的方向。
考虑到一种分子必须满足许多属性,例如溶解度和渗透性,才能成为药物样品[Bickerton等人 2012],另一个仍然存在的挑战是在生成模型期间同时优化生成的药物候选物的多个药物样性属性,同时保持其与目标蛋白质的结合亲和力。据我们所知,现有的工作在生成建模过程中没有明确优化这些属性。
8.4 分子和材料相互作用
作者:Zhao Xu,Limei Wang,Meng Liu,Montgomery Bohde,Yuchao Lin,Shuiwang Ji 推荐先决条件:第5.2节
本小节描述与分子和材料之间的相互作用相关的研究问题,包括预测分子-材料对结构的吸附能和每个原子的力(S2E和S2F),以及预测其弛豫最终结构和弛豫状态下的吸附能(IS2RS和IS2RE)。此外,我们在第8.4.5节讨论了分子-材料相互作用中未开发的生成任务。
8.4.1 问题设置。
假设分子-材料对中的原子总数为𝑛,匹配结构S表示为S = (𝒛,𝐶)。这里,𝒛 ∈ Z𝑛是原子类型向量,表示结构中所有𝑛个原子的原子类型(原子序数)。𝐶 = [𝒄1, ..., 𝒄𝑛 ] ∈ R3×𝑛是坐标矩阵,其中𝒄𝑖表示结构中第𝑖个原子的三维坐标。吸附能预测是引起研究界关注的第一个问题(S2E)。这个任务是学习一个函数𝑓𝐸,用于预测给定任意结构S的性质𝑒 ∈ R,其中𝑒是一个实数。第二个感兴趣的问题是在给定结构的原子类型和位置作为输入时预测每个原子的力(S2F)。在这里,目标是学习一个函数𝑓𝐹,用于预测任何给定结构S的力矩阵𝐹 ∈ R3×𝑛。每个原子的力驱动结构弛豫,直到分子-材料对结构达到其能量最低的弛豫状态。第三个问题旨在学习一个函数𝑓𝑅𝐸,根据其初始结构S𝑖𝑛𝑖𝑡作为输入来预测结构的吸附能𝑒𝑟𝑒𝑙 ∈ R,通常初始结构S𝑖𝑛𝑖𝑡是经验确定的。类似于IS2RE,最后一个问题IS2RS旨在学习一个函数𝑓𝑅𝑆,根据其初始结构作为输入来预测最终弛豫结构。在这个问题中,目标𝐶𝑟𝑒𝑙 ∈ R3×𝑛表示给定结构在其弛豫状态下的原子位置。Open Catalyst 2020(OC20)数据集 [Chanussot*等人 2021] 提供了吸附剂-催化剂对结构,并用于上述与分子-材料相互作用相关的问题的测试基准。
8.4.2 技术挑战。
对于上述不同的问题,存在不同的挑战。首先,对于能量预测问题(S2E),模型预测必须具有旋转不变性,因为能量是一个结构级的属性,对于分子-材料对结构的旋转是不变的。与能量不同,力预测问题(S2F)旨在预测每个原子的力矢量。因此,力的预测必须对结构的旋转具有等变性。类似地,弛豫能量预测问题(IS2RE)和弛豫结构预测问题(IS2RS)具有保持不变性和等变性的相同挑战。除了对称性,IS2RE和IS2RS问题还面临另一个挑战,即初始结构只提供了关于弛豫结构的粗略提示。因此,模型必须考虑结构弛豫,包括分子和材料原子,以获得准确的预测。
Table 31. 分子-材料对的能量、力和位置预测的现有方法比较。不同方法专注于不同的任务,并使用不同的流程来解决IS2RE和IS2RS任务。请注意,第5.2节介绍的方法也可以应用于本节介绍的任务。然而,出于简洁起见,此表仅包括几种最先进的方法,包括ForceNet [Hu等人 2021b],GNS+NoisyNode [Godwin等人 2022],Equiformer [Liao和Smidt 2023],EquiformerV2 [Liao等人 2023],SpinConv [Shuaibi等人 2021],GemNet-OC [Gasteiger等人 2022],SCN [Zitnick等人 2022]和eSCN [Passaro和Zitnick 2023]。

表格31提供了现有方法的摘要,包括它们的对称类型、网络顺序、任务和原始论文中使用的流程。正如在第5.2节中讨论的那样,既具有不变性又具有等变性的方法可以预测𝑆𝐸(3)-不变的能量𝑒。一旦能量被预测,可以使用公式𝐹 = −∂𝑒/∂𝒄𝑖来获得作用在原子上的𝑆𝑂(3)-等变力向量𝐹。对于不变模型(如SpinConv [Shuaibi等人 2021]和GemNet-OC [Gasteiger等人 2022])等模型,该公式是计算力的唯一选项。然而,等变方法也可以直接产生力。正如在第5.2节中讨论的,不变方法面临的一个主要挑战是效率问题。当将更多的几何信息(如角度和扭转角)纳入网络时,现有方法会受到高计算成本的困扰。另一方面,等变方法面临的一个主要挑战是,明确将物理约束条件纳入模型架构会限制网络的容量。因此,最近的研究试图设计既高效又具有表现力的GNN模型,而无需明确的物理约束。例如,ForceNet [Hu等人 2021b]以可扩展的方式直接使用原子坐标,但通过使用数据增强隐含地施加物理约束。
要解决IS2RE和IS2RS任务的挑战,其中只提供初始结构,需要考虑结构的松弛方法。主要有两种解决方案。第一种是使用经过良好训练的S2F模型来迭代地更新初始结构。原子位置根据当前结构的预测力逐步更新,直到预测的力接近零。虽然这种方法可以准确地模拟结构松弛,但需要大量步骤才能实现最终输出。最近,SCN [Zitnick等人 2022]和eSCN [Passaro和Zitnick 2023]都使用了这种间接方法,并指出它们大致等变,但阶数l > 1较高。结合eSCN和最近的直接方法Equiformer [Liao和Smidt 2023],EquiformerV2 [Liao等人 2023]在这些间接方法中实现了最先进的性能。它将eSCN卷积层应用于Equiformer网络结构,并使用几种附加技术,包括用于l = 0的独立S2激活和层归一化以及l > 0的矢量。与间接方法不同,直接方法旨在模拟初始和松弛结构之间的关系,其输出通常是差异𝐶𝑟𝑒𝑙 - 𝐶𝑖𝑛𝑖𝑡 [Godwin等人 2022]。因此,直接方法在训练和预测方面速度较快,但比间接方法精度较低。Equiformer [Liao和Smidt 2023],一种具有l = 1的注意力细节等变架构,在直接方法中实现了最先进的性能。目前,[Godwin等人 2022]提出的NoisyNode技术被广泛用于许多其他直接方法。然而,值得注意的是,GNS+NoisyNode [Godwin等人 2022]和ForceNet [Hu等人 2021b]都是基于GNS框架的,既不具有不变性也不具有等变性。这些方法将原子坐标作为输入,并使用旋转数据增强来提高性能。
8.4.4 数据集与基准
Open Catalyst 2020 (OC20) 数据集 [Chanussot* et al. 2021] 是测试机器学习模型在本小节描述的有关分子-材料相互作用问题上的有价值的资源。该数据集提供了吸附剂(分子)-催化剂(材料)配对结构,并包括三个任务,即结构到能量和力(S2EF)、初始结构到松弛能量(IS2RE)和初始结构到松弛结构(IS2RS),对应于第8.4.1节介绍的任务。
IS2RE 和 IS2RS 任务使用相同的数据集,其中包含输入的初始结构和输出的松弛结构和能量。该数据集最初分为训练、验证和测试集。训练集包含 460,328 个结构。对于验证和测试集,分为四个子集,即域内 (ID)、吸附剂域外 (OOD Ads)、催化剂域外 (OOD Cat) 和吸附剂催化剂域外 (OOD Both)。ID 集包含与训练相同分布的结构。OOD Ads 集包含具有未见过吸附剂的结构,OOD Cat 集包含具有未见过催化剂元素组成的结构,而 OOD Both 集包含具有未见过催化剂和吸附剂的结构。这四个验证集分别包含 24,943、24,961、24,963 和 24,987 个结构。测试集包含 24,948、24,930、24,965 和 24,985 个结构。请注意,测试集的标签不是公开的。因此,研究人员需要将他们的结果提交到Open Catalyst Project (OCP)的排行榜上,以在测试集上评估他们的模型。
与其他两个任务相比,S2EF 的数据集包含更多的结构。这是因为S2EF 数据集不仅包含初始和松弛结构,还包括弛豫轨迹中的中间结构等。该数据集包括 133,934,018 个用于训练的结构,而 ID、OOD Ads、OOD Cat 和OOD Both 验证集中分别包含 999,866、999,838、999,809 和 999,944 个结构。同样,ID 和OOD 测试集包括 999,736、999,859、999,826 和 999,973 个结构。
除了 OC20 [Chanussot* et al. 2021],最近还创建了 OC22 [Tran et al. 2023] 数据集,重点关注氧化物电催化剂。同样,它包括三个任务,即结构到总能量和力(S2EF-Total)、初始结构到总松弛能量(IS2RE-Total)和初始结构到松弛结构(IS2RS)。OC20 和 OC22 之间的一个主要区别在于 OC22 使用的是总能量目标,而不是吸附能目标。总的来说,这使得模型更通用,并能够计算更多的性质,但也更具挑战性。
8.4.5 开放研究方向
尽管在过去的几年里取得了显著的进展,但在建模分子-材料相互作用方面仍然存在一些挑战。首先,IS2RE/IS2RS 挑战中的松弛能量/结构不一定对应于分子-材料配对的全局最小吸附能量,并且对初始结构的更改非常敏感。最近,[Lan* et al. 2022] 使用了蛮力策略计算了许多初始配置中的松弛能量。为了有效地预测全局极小值,将需要使用更先进的策略来采样初始配置或构建不依赖于指定初始配置的模型。其次,尽管分子-材料相互作用中的许多材料是周期性的,但据我们所知,不存在任何模型在建模分子-材料配对时明确考虑这种周期性。相反,模型只考虑与小分子最近的晶胞。最后,目前的模型无法纳入系统的所有物理性质。许多模型通过包括分子动力学信息来提高性能,但当前的工作不能模拟诸如磁性或电荷效应等可以显著影响松弛能量/结构的性质。
生成分子-材料对:在这个领域的生成任务中,一个有前途的方向是在给定分子的条件下生成周期性材料。这个任务涉及训练一个生成模型,以在特定分子存在时产生具有特定属性(如适当的吸附能)的新周期性材料结构。为了完成这个任务,生成模型需要在包含不同吸附剂分子和它们对应的吸附能的周期性材料结构数据集上进行训练。这样的生成任务有许多潜在的应用,包括设计用于电催化剂的新材料。
尽管最近的研究已经探索了无条件材料生成,如第7.3节所述,但开发一个准确捕捉吸附剂分子和周期性材料之间复杂相互作用的生成模型具有挑战性。它需要对底层的物理学和化学有深刻的理解。而且,类似于第8.3节介绍的基于结构的药物设计任务,需要建模的庞大数据空间使得这个任务变得更加具有挑战性。此外,这个任务提出了一个独特的挑战,即分子的3D几何形状并不是静态的,将受到生成的材料分子的影响。因此,在生成以给定分子为条件的周期性材料时,生成模型需要考虑分子和材料之间的相互作用,包括分子几何形状的变化。