3 人工智能用于量子力学
在这一部分中,我们提供了有关如何设计高级深度学习方法以有效学习神经波函数的技术评述。在第3.1节中,我们概述了一般情况下定义和解决量子多体问题的方法。在第3.2节中,我们介绍了学习量子自旋系统基态的方法。在第3.3节中,我们介绍了学习多电子系统基态的方法。任务和代表性方法的概述如图9所示。
3.1 概述
作者:傅聪,张璇,徐胜龙,季水旺
量子力学是描述原子和亚原子粒子的物理规律的物理学分支[Feynman et al. 1965]。它对于解释微观领域中的量子系统的物理现象具有基本重要性,涵盖了从单个粒子到分子和材料的范围[Feynman et al. 2011; Griffiths and Schroeter 2018; Sakurai and Napolitano 2020]。量子态包含了有关量子系统的所有信息,并表示为波函数|𝜓〉。给定描述系统的一组变量,如其粒子的位置和动量作为输入,波函数|𝜓〉输出一个复数,表示系统测量的每个可能结果的概率幅度。波函数|𝜓〉是一个高维函数,需要指数级的信息来完全定义。获取量子系统的波函数是一个具有挑战性的问题,被称为量子多体问题。波函数|𝜓〉受薛定谔方程控制
𝐻ˆ|𝜓〉 = 𝐸|𝜓〉,(52)
其中𝐻ˆ是描述量子系统中粒子运动和相互作用的哈密顿算符,𝐸是该系统的总能量。在离散情况下,哈密顿算符𝐻ˆ可以表示为哈密顿矩阵𝐻。原则上,可以通过特征值分解获得𝐻的所有特征值和特征向量。然后,最小特征值是系统的基态能量,相应的特征向量称为基态,是最低能量的稳定态。在零温度下,基态完全确定了量子系统的所有性质。因此,我们关注如何获取给定量子系统的基态。
哈密顿矩阵的维度随着量子系统的大小(例如系统中的粒子数量)呈指数增长。例如,对于大小为𝑁的自旋系统,哈密顿矩阵的大小为2𝑁×2𝑁。因此,即使对于相对较小的系统,也不可行通过直接特征分解来获得基态。一种替代方法以近似方式获取基态及其能量是变分原理。考虑一个参数化函数|𝜓(𝜽)〉,表示一个量子态,其中𝜽是可学习参数。根据变分原理,|𝜓(𝜽)〉的能量必须大于或等于基态能量,即哈密顿矩阵的最小特征值。因此,通过𝜽近似基态,可以通过最小化状态的能量来优化变分参数𝜽。形式上,能量的期望值可以写成:

其中,𝐸0是基态能量,𝐸是与量子态|𝜓(𝜽)〉相关的能量。〈𝜓(𝜽)|是|𝜓(𝜽)〉的共轭转置,〈𝜓(𝜽)|𝜓(𝜽)〉表示这两个向量的点积。能量的期望值是数量𝐻ˆ𝜓(𝒔;𝜽)/𝜓(𝒔;𝜽)关于概率分布𝑝(𝒔) = |𝜓(𝒔;𝜽)|^2的均值,被称为局部能量。

图9. 人工智能在量子力学中的任务和方法概述。在本节中,我们重点关注两个子任务,包括学习自旋系统的基态和学习多电子系统的基态。用于学习自旋系统基态的方法根据它们用于表示量子态的网络类别进行分组。具体来说,Carleo和Troyer [2017],Gao和Duan [2017],Choo等人 [2018]和Chen等人 [2023]使用受限玻尔兹曼机。Cai和Liu [2018],Saito和Kato [2018],Saito [2018]和Saito [2017]使用前馈神经网络。Liang等人 [2018],Choo等人 [2019],Szabó和Castelnovo [2020]以及Fu等人 [2022c]使用卷积神经网络。Sharir等人 [2020],Hibat-Allah等人 [2020]和Luo等人 [2023a]使用自回归和循环神经网络。Yang等人 [2020]和Kochkov等人 [2021a]使用图神经网络。对于学习多电子系统的基态,一个重要的应用是分子。其中一类方法包括DeepWF [Han等人2019],PauliNet [Hermann等人2020],FermiNet [Pfau等人2020],FermiNet-GA [Lin等人2023b],FermiNet+SchNet [Gerard等人2022],PsiFormer [von Glehn等人2023],FermiNet+DMC [Ren等人2023; Wilson等人2021]和DiffVMC [Zhang等人2023c],旨在优化一次分子的单一几何结构。另一类方法包括DeepErwin [Scherbela等人2022],PESNet [Gao和Günnemann2021],PESNet++和PlaNet [Gao和Günnemann2023b],Globe [Gao和Günnemann2023a]和TAO [Scherbela等人2023],旨在同时优化同一分子或甚至不同分子之间的多个几何结构。除了分子,AGPs FermiNet [Lou等人2023]用于超流体。MP-NQS [Pescia等人2023],Cassella等人 [2023]和WAP-net [Wilson等人2023]用于均匀电子气。
局部能量的均值由于概率分布的高维性质而无法精确获得。相反,可以通过使用蒙特卡罗方法对概率分布进行采样来近似计算它。此外,梯度𝜕𝐸/𝜕𝜽也可以通过采样获得,并用于优化参数𝜽以降低能量。将这种将变分原理与蒙特卡罗采样相结合的方法称为变分蒙特卡罗(VMC),概述如图10所示。
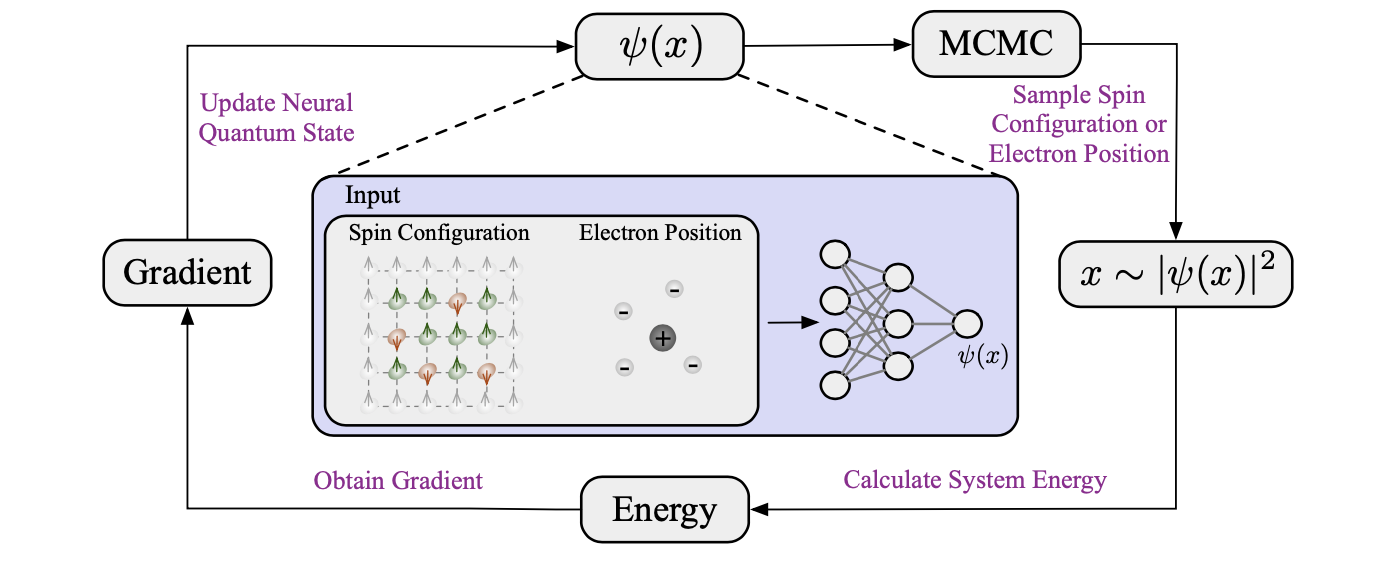
图10. 变分蒙特卡罗(VMC)的流程。神经量子态以自旋配置或电子位置作为输入,并输出波函数值。在VMC中,根据波函数确定的概率分布,使用马尔可夫链蒙特卡罗(MCMC)来采样自旋配置或电子位置。然后,从这些样本计算能量,并通过能量的梯度来更新神经量子态。
为了根据概率分布𝑝(𝒔)对输入配置进行采样,使用Metropolis-Hastings(MH)算法创建一个收敛到静止分布𝑝的输入配置的马尔可夫链。具体来说,对于马尔可夫链上的输入配置𝒔,根据提议分布𝑔(𝒔′|𝒔)提出一个新的输入配置𝒔′。然后,根据接受分布𝐴(𝒔′,𝒔)来接受或拒绝𝒔′。形式上,

如果𝒔′被拒绝,那么马尔可夫链上的下一个输入配置仍然是𝒔。一旦马尔可夫链收敛到静止分布,就可以从马尔可夫链中抽取样本,确保它们满足所需的分布。
在采样输入配置之后,我们可以将系统能量近似为局部能量的平均值,如下所示:

然后,我们可以使用能量𝐸作为损失函数,优化变分参数𝜽,使系统能量尽可能低。然后,具有最低能量的优化函数|𝜓(𝜽)〉可以被视为对基态的良好近似。在本节中,我们回顾了使用神经网络来表示量子态的方法,用于学习量子自旋系统和多电子系统的基态。
3.2 学习量子自旋系统的基态
作者:傅聪,季水旺
量子自旋模型是一种描述晶格上相互作用自旋的多体模型,这些自旋是紧密束缚在原子上的电子的自旋。这些自旋相互作用可以导致系统的各种磁性基态,如铁磁性、反铁磁性,甚至自旋液体,后者是一种异域的磁性状态,有望用于拓扑量子计算。了解量子自旋模型的基态为现代技术中不可或缺的磁性材料提供了宝贵的见解。
3.2.1 问题设置
在量子自旋系统中,每个自旋可以处于两种状态,自旋向上↑,自旋向下↓,或它们的叠加状态。𝑁个自旋的任何量子态都可以表示为2𝑁个自旋配置的叠加。所有自旋的组合构成了一个计算基础。具体来说,一个量子态可以写成:

其中|𝝈(𝑖)〉表示𝑁个自旋配置的数组,例如,↑↑↓...↓,而𝜓(𝝈(𝑖))是自旋配置|𝝈(𝑖)〉的波函数值。目标是使用神经网络来参数化波函数,并使用在第3.1节中描述的变分蒙特卡罗方法获得基态波函数。
3.2.2 技术挑战
学习量子自旋系统的基态面临着一些关键挑战,包括融合波函数的对称性、学习基态符号结构以及将方法扩展到不同的晶格几何形状。
保持对称性:在自旋系统中,学习的基态应满足一定的对称结构。量子自旋系统展现出丰富且有趣的对称性,这些对称性在传统的深度学习任务(如图像对象检测)中是不存在的。不同于图像,晶格是周期性的网格,具有额外的对称性,如旋转和反射,这些对称性可以归类为17个壁纸组,即使各种平面模式对应的变换都保持不变。尽管最强大的神经网络可以根据普适逼近定理从数据中自动学习这些对称性,但由于庞大的解空间和优化的难度,通常难以实现。将基态的对称性融入神经网络结构可以保证学得的基态也具有这些对称性,提高数据效率并有助于找到最优解。
学习符号结构:在量子力学中,波函数的符号结构通常指的是与量子态相关的复概率振幅的相位。学习基态的精确符号结构是一项具有挑战性的任务。有时,量子自旋系统的基态可能表现出严重的符号问题,其中自旋配置的微小变化可能导致波函数的符号变化,使得神经量子态难以收敛。这种现象在受挫折情况下更为严重,使神经网络难以捕捉基态的复杂符号结构。
多种几何形状:大多数现有方法仅适用于1D链或2D正方形晶格。然而,磁性材料的晶格几何形状可能比简单的正方形晶格丰富得多,并且对其基态以及磁性特性有显著影响。来自这种丰富几何形状的磁性受挫折为更多异域磁性特性的出现提供了可能。因此,将神经网络扩展到处理各种晶格几何形状至关重要。

表2. 不同研究在解决自旋系统的量子多体问题时如何应对几个挑战的总结,包括融合波函数的对称性、学习符号结构以及处理多种几何形状。为了考虑对称的基态结构,解决方案包括根据对称性对转换后的输入进行输出平均或使用群卷积。对于学习符号结构,解决方案包括使用复值网络隐式考虑相位、分别建模振幅和相位,或在某些特殊情况下将已知的Marshall符号规则作为参考符号结构。对于应用于多种几何形状,解决方案包括处理随机图和各种晶格几何形状。
3.2.3 现有方法
神经量子态(NQS)已经成为用于近似表示量子多体系统基态的强大变分方法。NQS可以根据神经网络类型分为五类,如图9所示。Carleo和Troyer [2017]提出了一项开创性工作,使用受限玻尔兹曼机(RBM)表示量子态。由于使用RBM作为变分方法的成功[Gao和Duan 2017; Deng等人 2017; Chen等人 2018a; Choo等人 2018; Chen等人 2023],研究人员开始探索更具表现力的深度学习方法来表示量子态,例如前馈神经网络[Cai和Liu 2018; Saito和Kato 2018; Saito 2018, 2017]。随后,卷积神经网络(CNNs)[Liang等人 2018; Choo等人 2019; Szabó和Castelnovo 2020]被应用于2D正方形晶格,并发现能够有效地表示高度纠缠的系统。然而,CNN不能自然地用于非网格晶格甚至随机图,这促使了对用于处理任意几何晶格的图神经网络(GNNs)[Yang等人 2020; Kochkov等人 2021a]的探索。此外,自回归和循环神经网络(RNNs)被用于表示量子态,使得可以直接采样自旋配置[Sharir等人 2020; Hibat-Allah等人 2020; Luo等人 2023a]。
除了不同的神经网络类型影响神经量子态的表达能力之外,各种方法还专注于解决上述提到的一些挑战,如表2所示。将基态的对称性融入神经网络中有助于减少假设空间。有效捕获波函数的符号结构对于神经量子态轻松收敛到最优解至关重要。此外,开发单一的神经量子态,可以在多个晶格上运行,可以显著提高其实用性和多功能性。
保持对称性:为了捕捉基态的对称性,大多数工作[Nomura和Imada 2021; Nomura 2021; Ferrari等人 2019; Choo等人 2018, 2019; Chen等人 2023]使用对称性平均技术,该技术涉及根据对称性群变换来转换输入,然后将每个输出的平均值作为最终预测的基态值。另一种保持对称性的方法是使用[Cohen和Welling 2016]提出的群等变卷积。在GCNN [Roth和MacDonald 2021]中,作者提出了一个通用框架,使用群等变卷积考虑完整的壁纸组,并展示了GCNN在正方形和三角形晶格上的有效性。通过在隐藏层之间屏蔽一些过滤器,可以将GCNN映射到对称性平均模型中。
学习符号结构:除了捕捉基态的振幅之外,符号结构的学习也至关重要。一些工作通过使用具有复值参数的单一神经网络同时学习振幅和相位[Carleo和Troyer 2017; Choo等人 2019; Sharir等人 2020]来学习振幅和相位。Choo等人[2019]将Marshall符号规则作为参考符号结构并将其纳入网络设计中。Marshall符号规则提供了一个简单的符号结构,对于某些极限情况下的二部图,如𝐽1 = 0或𝐽2 = 0的𝐽1 − 𝐽2海森伯模型,已知。但是,对于更复杂受挫折的基态,没有这样简单的先验符号结构可供使用。Cai和Liu [2018]将前馈神经网络修改为两个分支,分别预测基态的振幅和符号,然后将它们相乘。他们使用余弦函数作为预测符号的激活函数,适合捕捉输入自旋的振荡特性。Kochkov等人[2021a]分别预测波函数的对数振幅和相位,并表明直接预测相位可以有效地推广学得的符号结构。Szabó和Castelnovo [2020]使用两个实值神经网络来建模振幅和符号结构。具体来说,他们通过对每个局部自旋的预测矢量和求和来计算全局相位。此外,他们采用了两阶段优化方法。首先,他们保持所有自旋配置的波函数振幅相同,并仅优化相位以最小化系统能量。在这个阶段中,可以提供良好的初始符号结构,因为最优符号结构对振幅的依赖较弱[Szabó和Castelnovo 2020; Marshall 1955]。然后,在第二阶段同时优化符号结构和振幅。
多种几何形状:上述大多数工作仅使用正方形晶格作为测试基础。实际有用的波函数假设应该适用于不同的晶格几何形状并表现良好。GNA [Yang等人 2020]提出了通用波函数假设,并在2D Kagome晶格、三角晶格和随机连接图上进行了实验。Kochkov等人[2021a]设计了另一种基于GNN的假设,使用子晶格编码来表示节点在尊重晶格对称性的单元格中的位置。除了使用GNN实现在任意晶格上的适用性之外,LCN [Fu等人 2022c]提出了使用虚拟顶点进行网格卷积的方法,以扩展原始晶格并将其转换为正方形晶格,从而可以应用常规CNN。
3.2.4 优化方法
有几种方法可以优化神经网络量子态。直接方法是将系统能量直接计算为损失函数,并使用深度学习中的梯度下降方法,如SGD和Adam,来更新网络参数[Roth和MacDonald 2021; Fu等人 2022c]。能量梯度为

其中𝑂𝑘 = 𝜕𝜓(𝝈;𝜽)/𝜕𝜃𝑘是关于第k个网络参数的变分导数,而𝑂是𝑂𝑘的复共轭。𝐸𝑙𝑜𝑐 =
Í 𝜓(𝝈(𝑗);𝜽) 𝐻𝑖 𝑗 (𝑖) 是关于
∗
𝜓(𝝈;𝜽)的局部能量。为了从由波函数𝜓定义的所需概率分布𝑝(𝝈) = Í𝑖 |𝜓(𝝈(𝑖))|2 中采样自旋配置,我们可以使用第3.1节中描述的马尔科夫链蒙特卡洛(MCMC)方法。对于自旋系统,马尔科夫链中提出的自旋配置可以通过在晶格中随机翻转一个自旋来获得。因此,建议概率是对称的,即𝑔(𝝈′|𝝈) = 𝑔(𝝈|𝝈′)。因此,接受概率可以简化如下:

优化神经网络量子态的另一种方法是使用随机重构(SR) [Sorella等人 2007],它表示了在变分空间中的虚时间演化过程。当一个量子态经历虚时间演化时,最终会收敛到系统的基态。在随机重构中,网络参数被更新为:

在其中,𝜂是学习率,𝚫𝑬是能量梯度,𝑆𝑖𝑗 = 〈𝑂𝑖∗𝑂𝑗〉 − 〈𝑂𝑖∗〉〈𝑂𝑗〉。与梯度下降的唯一区别是协方差矩阵𝑆的存在。随机重构通常更加健壮,对学习率的敏感性较低。但是,SR的局限性在于矩阵𝑆的大小等于神经网络参数的数量,因此在具有大参数的神经网络上计算其逆矩阵在计算上具有较高的代价。Kochkov和Clark [2018]提出的另一种可以克服SR局限性的优化方法是虚时间监督波函数优化(IT-SWO)。IT-SWO在能量梯度和随机重构方法之间进行插值优化[ Kochkov等人2021a]。它通过优化波函数模型来最大化当前变分状态𝜓(𝝈;𝒘)与虚时间演化状态(𝐼 − 𝛽𝐻)𝜓 (𝝈; 𝒓)之间的重叠度,其中𝒘和𝒓表示当前状态的参数和上次优化迭代结束时的状态。在每次优化迭代中,IT-SWO首先更新目标状态并在当前迭代期间保持其不变,然后通过随机梯度下降执行多个内部步骤以更新当前状态。
3.2.5 数据集和基准。
与传统的机器学习任务不同,用于确定量子自旋系统基态的模型不能在现有数据集上进行训练。相反,该模型针对特定的量子自旋系统进行训练,该系统由晶格和哈密顿量定义。在训练过程的每个步骤中,数据是从量子系统的波函数(神经网络)中动态采样的。这种方法被E等人[2020]描述为并行机器学习。通常考虑各种晶格系统,如正方形、蜂窝、三角形和卡戈梅晶格。最常用的哈密顿量是𝐽1-𝐽2量子海森堡模型,这是研究量子材料磁性性质的原型模型。在评估指标方面,系统的能量用作近似基态与真实基态的接近程度的度量。较低的能量表示更准确的近似。
3.2.6 开放研究方向。
神经网络量子态已经显示出在表示量子自旋系统的基态方面具有潜力,但仍然需要进一步探索一些挑战。首先,我们需要更具表现力和高效的波函数模型,以便在任意大的系统中扩展,以便在实际应用中有用。随着系统的规模增加,自旋相互作用的复杂性也增加。其次,设计具有可证明足够表现力的神经波函数仍然是一个开放问题,特别是对于表现出高度受挫和强相关性的量子系统。第三,费米子晶格系统具有波函数的独特反对称性质,这使得神经量子态难以有效地编码这种对称性。最后,在变分蒙特卡洛中,通常使用马尔科夫链蒙特卡洛(MCMC)来从由波函数确定的概率分布中采样自旋配置,然后计算系统能量。然而,使用MCMC进行精确采样是困难的,样本可能仍然存在相关性,导致能量估计不准确。通常,在两个样本之间添加N个退火MCMC步骤,其中N代表系统的大小,以减小样本之间的自相关性。但是,这使得对于更大的系统来说,采样过程在计算上变得昂贵。在[Sharir等人2020]中提出了解决这一挑战的潜在方法。他们使用自回归模型来表示量子态,从而避免了MCMC采样,可以支持更高效和精确的采样。
3.3 学习多电子系统的基态
作者:Xuan Zhang,Nicholas Gao,Stephan Günnemann,Shuiwang Ji
与量子自旋系统相比,量子力学中更一般的情景是粒子可以在空间中自由移动,例如分子和固体中的电子运动。研究多电子系统是量子化学的核心,其中分子的性质是基于量子物理学的第一原理直接计算的。具体来说,准确描述分子的基态非常重要,因为基态决定了分子最稳定的状态,对于理解其结构和化学性质非常重要。与量子自旋系统相比,电子的自旋在哈密顿量中不出现,因此可以事先固定[Foulkes等人,2001]。因此,波函数仅作用于每个电子的R3中的空间坐标。然而,适当波函数的搜索空间随着电子数量𝑁的增加呈指数级增长。此外,电子的费米特性显着增加了问题的难度[Ceperley,1991]。例如,已经表明,由于Pauli排斥,费米子可能会出现的符号问题的解决是NP难的[Troyer和Wiese,2005]。在本节中,我们主要讨论使用量子蒙特卡罗(QMC)方法学习分子的基态。此外,类似的方法也已应用于超流体和均匀电子气(HEG),为了完整起见,我们也将简要回顾这些方法。
3.3.1 问题设置。
分子由电子和原子核组成。在Born-Oppenheimer近似[Born和Oppenheimer,1927]中,原子核被视为固定粒子,因此量子态完全由电子的自旋和三维坐标决定。在基态,电子的自旋可以通过化学规则确定,例如Aufbau原理、Hund规则和Pauli排斥原理。因此,我们能够仅根据电子坐标来定义波函数。形式上,给定𝑁↑个自旋向上的电子,𝑁↓个自旋向下的电子。它们的3D笛卡尔坐标集被定义为𝒓 = [𝒓1,...,𝒓𝑁↑+𝑁↓] ∈ R(𝑁↑+𝑁↓)×3,其中前𝑁↑个电子具有自旋向上,后𝑁↓个电子具有自旋向下。波函数𝜓:R(𝑁↑+𝑁↓)×3 → R将坐标集映射到标量值。在连续情况下,哈密顿算子𝐻ˆ:(R(𝑁↑+𝑁↓)×3 → R) → (R(𝑁↑+𝑁↓)×3 → R)是一个函数,将一个波函数映射到另一个函数,定义了分子的能量,定义为

其中第一项代表动能,第二项代表库伦势能,定义为

因此,可以事先确定电子的自旋。给定哈密顿算符,局部能量 𝐸(𝒓) = [𝐻ˆ𝜓](𝒓) 可以根据波函数 𝜓(𝒓) 计算得到。

在一个多电子系统中,一个基本的约束是其波函数必须在两个具有相同自旋的电子置换时是反对称的,这个概念源自于泡利不相容原理。在量子力学中,交换两个不可区分的粒子不会影响粒子的概率密度。在我们的情况下,如果两个电子具有相同的自旋,它们就无法区分。因此,对于任何具有相同自旋的(𝑖,𝑗),𝜓(...,𝒓𝑖,...,𝒓𝑗,...)2 = 𝜓(...,𝒓𝑗,...,𝒓𝑖,...)2。此外,根据它们的交换对称性[Feynman et al. 1965],不可区分的粒子分为玻色子,如光子,和费米子,如电子,它们的交换对称性决定了波函数 𝜓 在交换两个粒子的位置时是否保持不变或改变符号。电子是费米子,因此波函数在两个具有相同自旋的电子置换时必须是反对称的,即 𝜓(...,𝒓𝑖,...,𝒓𝑗,...) = -𝜓(...,𝒓𝑗,...,𝒓𝑖,...)。这种反对称性导致了费米-狄拉克统计的粒子分布,从而从根本上改变了费米子的行为。因此,寻找基态可以被形式化为一个受约束的优化问题。在变分蒙特卡洛的上下文中,波函数 𝜓 被参数化的函数类 𝜓𝜃 来近似。在这种情况下,学习基态等价于以下优化问题:

在上面的优化目标中,能量期望值是通过蒙特卡洛采样获得的样本的平均值来计算的。根据前面提到的变分原理(方程(53)),任何波函数的能量期望值都保证大于基态能量,当 𝜓𝜃 收敛到基态波函数时,下界得以实现。
上面的公式为获得单一分子的基态能量提供了一个框架。在本节中,我们还考虑了同时优化多种几何设置的情况。例如,通常我们有兴趣研究分子结构变化所导致的能量变化。联合优化通过消除了需要针对每个核心配置重新优化的需求,提高了计算效率。形式上,我们将势能面(PES)定义为一个函数 𝐸: M → R,其中 M = {𝑀 = {𝒄𝑖,𝑧𝑖}|𝑀|, ci ∈ R³, 𝑧𝑖 ∈ Z} 是 𝑖=1 可能分子的集合,从分子结构(核的坐标和电荷)到能量的映射。经典地,要获得 PES,需要多次重复单一结构的计算。基于基于神经网络的解决方案的出现,现在可以使用单一模型来模拟 PES 的自然生成的解。具体来说,在这种情况下,人们有兴趣寻找一个波函数 𝜓𝜽 : R(𝑁↑+𝑁↓)×3 × M → R,其中波函数现在还依赖于分子结构,除了电子坐标。按照 Gao 和 Günnemann [2023a],我们将这样的 𝜓𝜽 称为广义波函数。请注意,这个公式不应与没有Born-Oppenheimer近似的薛定谔方程混淆,其中核被视为波动并因此被视为波函数的一部分。在这里不是这种情况,我们仍然只模拟电子波函数,但将波函数条件付在分子结构上。最后,使用广义波函数,可以导出势能面 𝐸(𝑀) = ∫ 𝜓𝜽(𝒓, 𝑀)𝐻ˆ𝑀𝜓𝜽(𝒓, 𝑀)𝑑𝒓,其中 𝐻ˆ𝑀 指的是分子 𝑀 的哈密顿量。
此外,超流体和均匀电子气也可以被建模为连续空间中的费米子。然而,这些问题不涉及核并且在哈密顿量中使用不同的势能。另一个主要的区别是这些问题在空间中是周期性的。尽管如此,对于单分子设置的一般方法仍然可以应用。
在我们的讨论中,波函数直接以连续电子坐标作为输入,因此反对称约束必须明确编码在 𝜓𝜃 中。具有这种连续表示的方法称为第一量子化方法。另一种选择是在第二量子化方法中[Barrett et al. 2022; Choo et al. 2020; Herzog et al. 2022],波函数是在反对称函数的离散基础上表示的。关于第一量子化和第二量子化之间的比较在 Hermann et al. [2022] 中有讨论。最近,第一量子化方法因其在反对称基础选择以外的灵活性而变得更受欢迎。此外,为了更全面地描述多电子系统,可以使用与地面状态类似的方法来研究激发态[Entwistle et al. 2023; Feldt and Filippi 2020]。然而,这些细节不在本节讨论的范围内。
有许多与使用QMC找到多电子基态相关的挑战,包括满足费米子反对称性约束、为单个电子(轨道)设计富有表现力的神经网络、实现良好的优化,以及有效地学习多个几何结构的广义波函数以提高计算效率。
费米子反对称性:正如在第3.3.1节中介绍的那样,费米子反对称性是量子物理强加的一个严格约束,电子的神经波函数必须严格遵守它。未能编码反对称性约束将使变分保证失效,并导致不符合物理规律的更低能量。尽管深度神经网络可以逼近任意复杂的函数,但施加这种硬约束带来了独特的挑战。
轨道建模:电子通过库仑势和泡利排斥相互作用,这可以导致波函数中高度非线性的地形。因此,神经网络必须具有强大的能力来建模每个电子(称为轨道)的波函数,同时考虑与其他电子的相互作用。此外,量子物理为我们提供了一些系统的先验知识,可能难以直接用神经网络建模。因此,在轨道建模中融入物理知识对于获得符合物理规律的解非常重要。
优化:尽管原则上我们可以通过VMC获得任意的逼近精度,但要实现对神经波函数朝向基态的有效优化是具有挑战性的。这部分是由于问题对高精度的要求。化学精度被定义为1千卡/摩尔(1.594 mEh或0.043电子伏特)[Pfau et al. 2020],与总能量相比非常小。例如,对于N2分子,能量估计的误差必须低于0.2%才能用于化学应用[Gerard et al. 2022]。因此,有效的优化方法对于获得准确且稳定的优化结果至关重要。
多种几何结构:在多种几何结构设置下存在一些独特的挑战。首先,需要特殊考虑,以使学习的波函数能够适应各种分子构型,包括不同的原子核位置和可变数量的核和电子(例如离子体系),同时遵守费米子反对称性。其次,作为可观测度量的PES 𝐸 对于欧几里得群 𝐸(3) 即平移、旋转和反射以及置换群 𝑆𝑀 是不变的。然而,作为一个抽象概念,电子波函数并不表现出这种对称性。因此,挑战在于设计导致不变能量的广义波函数。可以证明,要获得这样的行为,需要设计破对称性协变波函数。第三,先验知识为我们提供了关于极限行为的附加约束。其中一种属性是大小一致性,即复制的非相互作用系统的能量是单个系统能量的两倍。将这些行为实施到波函数中仍然是一个减少函数搜索空间的挑战。最后,虽然波函数与能量直接相关,但从波函数中获取能量仍然很昂贵,因为它需要数值积分。近似推理方法承诺加速该过程并实现高分辨率的PES。
现有的方法表明,基于VMC的神经网络在建模多电子系统的基态方面具有强大的能力。传统方法,如DFT或CCSD(T),在强关联设置下,例如键断裂时,要么导致不可靠的结果,要么随着系统大小的增加而不利。VMC与深度神经网络相结合已经显示出能够胜过传统方法[Pfau et al. 2020; Gerard et al. 2022]。特别是,尽管DFT比深度VMC方法更具扩展性(𝑂(𝑁3)与𝑂(𝑁4)),但深度VMC方法可以实现更高的精度。另一方面,深度VMC方法比CCSD(T)更快,可以实现相似或更高的精度。此外,尽管CCSD(T)可以应用于较大的分子,但必须选择较小的基组。接下来,我们简要介绍现有方法是如何解决第3.3.2节中列出的挑战的。首先,我们描述如何在网络中编码费米子反对称性,特别是使用Slater行列式。接下来,我们描述现有方法中网络的设计。有了这两个组件,我们已经可以有一个工作的神经波函数模型。然后,我们讨论如何有效地优化网络以达到基态。最后,我们描述通过广义波函数重用和加速计算的策略。这些挑战和现有方法在表3中总结。
费米子反对称性:为了设计满足费米子反对称性(方程(65))的波函数,一个已经建立的方法是Slater行列式[Slater 1929]。Slater行列式波函数是通过计算一个矩阵的行列式来构建的,该矩阵由将𝑁分子轨道函数应用于每个𝑁电子中的一个,以便矩阵的每一行都编码一个电子。其关键动机是,当两个电子互换时,矩阵中的两个对应行也会互换,因此其行列式将改变符号。形式上,让 𝝓↑ 和 𝝓↓:R3×R(𝑁↑+𝑁↓)×3 →R𝑁↑+𝑁↓ 为两个单轨道函数,它们将3D电子坐标映射到(𝑁↑+𝑁↓)-维特征向量,其中 𝝓↑ 用于编码自旋向上的电子,𝝓↓ 用于编码自旋向下的电子。 𝝓↑ 和 𝝓↓ 都接受一个电子坐标以及所有电子坐标作为输入,并产生一个(𝑁↑+𝑁↓)-维向量。单轨道函数的目标是为每个电子生成一个嵌入,而来自所有电子的信息则用于提供上下文信息。在使用 𝝓↑ 或 𝝓↓ 对每个电子进行编码后,会得到一组包含𝑁↑+𝑁↓个元素的特征向量,每个特征向量都包含𝑁↑+𝑁↓个元素。这些特征被堆叠成一个矩阵,其中每一行表示一个电子。Slater行列式波函数 𝜓 是:
\[𝜓(𝒓) = det(𝝓↑(𝒓1), 𝝓↑(𝒓2),..., 𝝓↑(𝒓𝑁↑); 𝝓↓(𝒓1), 𝝓↓(𝒓2),..., 𝝓↓(𝒓𝑁↓))\]
这里𝒓是电子的空间坐标向量。Slater行列式是费米子反对称的,因为当任意两个电子的坐标交换时,行列式的值会发生变化。因此,它是一种有效的方法来满足费米子反对称性。
表3. 总结了学习多电子基态的挑战和现有方法。对于电子,由于量子物理规定的费米子反对称性,存在特殊挑战。大多数现有的波函数模型通过Slater行列式来解决这一挑战,但它们在模拟轨道函数方面具有不同的网络设计。此外,为了使学习准确和实用,实现有效的优化至关重要。最后,分子的多样性和灵活性要求采用多几何处理方法,以提高计算效率。

然后计算该矩阵的行列式:

请注意,自旋向上和自旋向下的电子的轨道函数是不同的,因此反对称性只在它们之间进行交换时才存在。
例如,当 𝑁 ↑ = 2 和 𝑁 ↓ = 1(锂原子)时,通过示例中的情况可看出,当 𝒓2 和 𝒓3 交换时,费米子反对称性得到了满足:

为了进一步增加表达能力,可以计算多个 Slater 行列式,每个行列式都使用不同的轨道函数集合,最终的波函数是 Slater 行列式的线性组合。当使用 𝑘 个 Slater 行列式时,波函数可以表示为如下形式:

除了 Slater 行列式之外,还有其他方法可以实现反对称性。DeepWF [Han et al. 2019] 和 Pang 等人 [2022] 提出将反对称性强加给每对电子。波函数的定义形式为 Î𝑖<𝑗 (𝑓 (𝒓𝑖;𝒓) − 𝑓 (𝒓𝑗;𝒓)),其中 𝑓 输出标量值。当一对 (𝒓𝑖, 𝒓𝑗) 被交换时,乘积的符号会改变。这种策略被证明是 Slater 行列式的特殊情况(可以写成 Vandermonde 矩阵的行列式 [Pang et al. 2022]),但计算成本较低。Lin 等人 [2023b] 推广了对电子所有可能的排列的总乘积行列式计算,最终波函数是所有排列结果的总和,即 Í𝜋 sign(𝜋)𝑔(𝜋(𝒓)),其中 𝜋 遍历所有排列,sign(𝜋) 给出每个排列的符号,而 𝑔 是一个将排列后的 𝒓 映射到标量的函数。然而,这会导致阶乘复杂度。最后,反对称 geminal 动力学(AGP)波函数在用神经网络波函数建模超流体时表现出色 [Lou et al. 2023]。在这里,使用成对的轨道函数 𝜙 : R3 × R3 × R(𝑁↑×𝑁↓)×3 → R,并将波函数构造为 𝜓(𝒓)=detΦ,其中Φ𝑖,𝑗 =𝜙(𝑟𝑖,𝑟𝑗,𝒓),𝑖∈{1,...,𝑁↑},𝑗∈{𝑁↑+1,...,𝑁↑+𝑁↓}。

图 11. 多电子波函数计算的流程,以分子为例进行了说明。网络的输入是一组带有自旋向上的 𝑁 ↑ 电子和自旋向下的 𝑁 ↓ 电子的 3D 电子坐标。自旋结构(↑ 或 ↓)以及原子核的位置是固定的。神经网络用于为每个电子生成 𝑘 (𝑁 ↑ + 𝑁 ↓)-维特征向量,然后将它们级联成 𝑘 (𝑁 ↑ + 𝑁 ↓) × (𝑁 ↑ + 𝑁 ↓) 的矩阵。最后,计算这些矩阵的行列式,并对它们进行线性组合,得到最终的波函数值。
轨道建模:虽然 Slater 行列式解决了多电子系统的交换反对称性,但它并不能保证优化波函数的准确性。为了准确地建模基态波函数,轨道函数 𝝓↑ 和 𝝓↓ 必须来自于一个灵活的函数家族。经典上,轨道函数被建模为来自单粒子薛定谔方程解的单粒子轨道。FermiNet [Pfau et al. 2020] 和 PauliNet [Hermann et al. 2020] 成功地使用神经网络来建模轨道函数,同时使用 Slater 行列式作为反对称聚合,其中所有 3D 电子坐标首先被编码为 (𝑁 ↑ + 𝑁 ↓) × 𝑘 维特征向量,使用置换等变神经网络 𝝓↑ 和 𝝓↓。然后,这些向量被级联成 𝑘 (𝑁 ↑ + 𝑁 ↓) × (𝑁 ↑ + 𝑁 ↓) 的矩阵 𝜃𝜃(例如,𝑘 = 16)。最后,计算这些矩阵的行列式,并且最终波函数值是行列式的线性组合。因此,网络中的参数是轨道网络的参数以及行列式的组合权重。为了构建量子态的完整表示,必须考虑所有粒子之间的所有成对信息,包括电子和原子核。为此,常用的输入特征是电子-电子和电子-原子核对之间的相对向量和距离。另外,还可以将称为 Jastrow 因子的对称部分 𝑒𝐽 (𝒓;𝜽 ) 乘以最终波函数。
单电子特征计算:为了捕捉复杂的物理相互作用,轨道网络 𝝓↑ 和 𝝓↓ 必须将所有电子的位置集体考虑在内。因此,在编码一个电子时,所有其他电子也必须被编码以提供上下文信息。因此,𝝓↑ 和 𝝓↓ 必须能够从其他电子中收集信息。
PauliNet [Hermann et al. 2020] 使用基于距离的卷积来收集来自邻近电子的信息,类似于 SchNet [Schütt et al. 2018],其中卷积权重是基于相对距离计算的:𝒉′ =Í 𝒘(∥𝒓 −𝒓 ∥ ;𝜽)⊙𝒇(𝒉 ),其中 ⊙ 表示逐元素相乘,𝒘 对不同自旋的情况计算不同。同样的计算也应用于原子核。FermiNet [Pfau et al. 2020] 利用有关电子结构的均场信息以及每对电子之间的距离特征。具体来说,FermiNet 维护两个计算分支,分别计算单粒子特征和成对特征。在每一层,单粒子特征 𝒉𝑖 和成对特征 𝒉𝑖 𝑗 在电子上平均以获得每个自旋的全局表示,然后将它们级联到下一层的单粒子特征中,用于下一层 𝒉𝑖′ =𝒇([𝒉𝑖,Í𝑗,𝜎(𝑗)=↑ 𝒉𝑗,Í𝑗,𝜎(𝑗)=↓ 𝒉𝑗,Í𝑗,𝜎(𝑗)=↑ 𝒉𝑖𝑗,Í𝑗,𝜎(𝑗)=↓ 𝒉𝑖𝑗]),其中 𝜎 (𝑖) 表示第 𝑖 电子的自旋,[·] 表示级联。FermiNet+SchNet [Gerard et al. 2022] 将 FermiNet 中的简单交互替换为从 PauliNet 那里积分的卷积。通过放弃 FermiNet 的均场,PsiFormer [von Glehn et al. 2023] 利用注意机制捕获了所有成对电子-电子相互作用,从而获得了明显更低的能量。相比之下,Moon [Gao and Günnemann 2023a] 通过使用连续卷积将 FermiNet 的全局均场替换为原子核上的局部均场,结果获得了与 PsiFormer 类似的准确性。对于超流体和均匀电子气,可以使用类似的网络,但必须使用周期函数将输入坐标嵌入以尊重问题的周期性 [Pescia et al. 2022; Cassella et al. 2023]。MP-NQS [Pescia et al. 2023] 使用消息传递网络来建模均匀电子气的量子态。
费米子反对称性不要求 𝝓𝜽↑ 和 𝝓𝜽↓ 是相同的映射。因此,在最一般的设置中,它们应该能够生成不同的计算。虽然传统上选择 𝝓↑ 和 𝝓↓ 为不同的函数可以更好地获得变分能量,但对于自旋匹配的系统的基态,即所有电子自旋都成对时,𝝓↑ = 𝝓↓。在神经网络设置下,Gao and Günnemann [2023b] 已经表明,对于自旋匹配的系统,实施这种约束可以提高能量并加速神经网络基础的波函数的优化。然而,在实际应用中,这两个网络的大多数参数可以共享。具体来说,在网络的每一层,每个电子都被编码为特征 𝒉 ∈ R𝑑,其中 𝑖 𝑑 是隐藏维度。下一层的特征是通过公式 𝒉𝑖′ = 𝒇(𝒉𝑖,𝒉𝜎,{𝒉𝑗}𝑗≠𝑖) 计算的,其中 𝒉𝜎 , 𝜎 ∈ {↑, ↓} 是一个特征,用于区分不同自旋的全局信息,区分自旋向上和自旋向下的电子,例如,𝒉↑ 和 𝒉↓ 可以分别计算为自旋向上电子特征和自旋向:

继续优化的另一个重要组成部分是优化器的选择。二阶优化器,如自然梯度,被发现对于实现准确的优化至关重要。与直接沿着梯度的反向方向更新参数的一阶方法相比,自然梯度通过使用 Fisher 信息矩阵的逆对梯度进行预处理,以便在分布方面进行最陡方向的更新。具体来说,参数的更新为𝜽 ←𝜽−𝜂𝐹−1∇𝜽L(𝒓;𝜽)。当处理未归一化的波函数时,参数更新等效于随机重配置,其中𝐹 ∝E O−E [O]O−E [O],其中 O =𝜕log|𝜓𝜽(𝒓)|。然而,对于大型模型来说,计算 𝐹 −1 是不可行的。因此需要近似。通过对 Fisher 矩阵进行某些假设,KFAC [Martens and Grosse 2015] 通过克罗内克积来分解 Fishier 矩阵以加速计算。或者,可以使用共轭梯度 (CG) 方法来近似 𝐹−1∇𝜽 L(𝒓;𝜽) [Gao and Günnemann 2021]。
除了 VMC,还存在其他优化神经波函数的方法。在扩散蒙特卡洛 (DMC) 中,每个样本额外分配一个权重,以使加权平均的采样能量更接近真实基态能量。为了实现这一点,权重是基于虚时间演化计算的。在 DMC 中,采样是通过 Langevin 动力学进行的,其中样本是根据量子漂移 (或机器学习中的分数) 生成的,分数定义为 ∇𝒓 log𝜓。该过程近似于虚时间演化算符的迭代应用𝜓 ← 𝑒 −𝜏 𝐻ˆ 𝜓,其中 𝜏 是演化时间。分数为每个电子提供一个三维向量,该向量指向更高概率密度的方向。Ren 等人 [2023];Wilson 等人 [2021] 首先使用 VMC 训练 FermiNet,然后使用 DMC 进一步接近基态。此外,DiffVMC [Zhang 等人 2023c] 通过直接参数化分数来结合 VMC 和 DMC。与更新样本权重不同,Diff VMC 通过基于分数匹配的特殊设计的损失函数直接更新参数化的分数函数 [Hyvärinen and Dayan 2005]。
多种几何形态的泛化波函数:学习泛化波函数,无论是覆盖给定分子的完整势能面还是跨不同化合物,都面临着各种挑战,这些挑战不适用于单个结构的计算。
多种几何形态 — 泛化轨道:在学习泛化波函数时,一个关键挑战是将分子轨道函数 𝜙𝑖 调整到分子结构。已经有各种方法来实现这一目标。Scherbela 等人的首个作品 [2022](DeepErwin)通过在不同结构之间共享大多数参数,并仅为每个结构重新训练特定权重来解决此问题。与此同时,Gao 和 Günnemann [2021] 提出了一种称为“势能面网络”(PESNet) 的方法,采用两层网络方法来适应轨道函数。在 PESNet 中,轨道函数由另一个仅作用于原子核的神经网络参数化,类似于监督替代模型。这避免了完全重新训练的需求,只需优化一次,即可对分子的整个势能面进行建模。然而,虽然权重共享方法可以转移到不同的原子集合,但 PESNet 没有这样的能力。
当学习不同化合物的泛化轨道时,即不同数量的原子集合,分子轨道函数参数化的问题 {𝜙 }𝑁 的问题受到不同数量的分子轨道 𝑁 影响,这与电子数量相对应。尽管许多权重仍然可以共享,但仍然需要为每种分子单独优化波函数[Scherbela et al. 2022]。有两项并行工作解决了这个问题并避免了个体优化:可传递的原子轨道 (TAOs) [Scherbela et al. 2023] 和图学习轨道嵌入 (Globe) [Gao and Günnemann 2023a]。在 TAO 中,分子轨道函数
多种几何形态 — 对称性:由于能量 𝐸 是可观察的,它在欧几里得群 𝐸(3) 方面是不变的。具体来说,让 𝑈𝑅 是与旋转矩阵 𝑅 相关的幺正算符,旋转后的薛定谔方程为 𝑈 𝐻ˆ𝑈†𝜓 = 𝐸𝜓,其中 𝑈 𝐻ˆ𝑈† = −1 Í ∇2+Í 1 − 𝑅 𝑅 𝑅 𝑅 2 𝑖 𝑖 𝑖<𝑗∥𝒓𝑖−𝒓𝑗∥
Í 𝑧𝐼 + Í 1 为旋转后的哈密顿算符。从这个公式中,可以看出旋转哈密顿算符会解决旋转后的薛定谔方程,即:
𝑈 𝐻ˆ𝑈†𝜓 = 𝐸𝜓
这意味着对于给定系统,旋转一个本征函数 𝜓 会得到相同能量的本征函数,即旋转操作保持系统的能量不变。
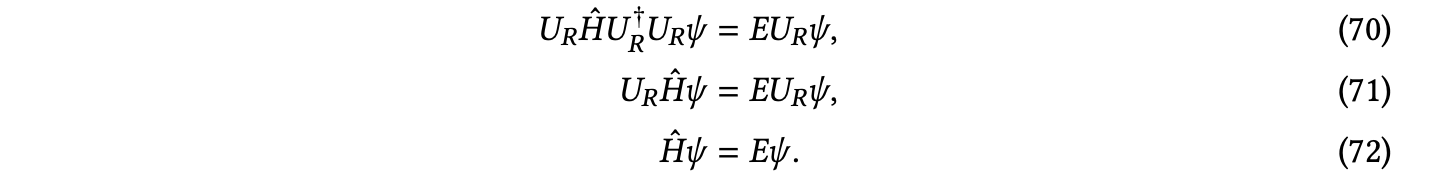
因此,为了获得不变的能量,必须具有等变的波函数。一个简单的实现方式是像 PauliNet [Hermann et al. 2020] 那样使用不变的波函数,但由于波函数不必具有不变性,这严重限制了函数类别,通常会导致更高的能量。相反,当前的方法要么依赖于使用随核结构旋转的等变坐标框架 [Gao and Günnemann 2021; Gao et al. 2022; Gao and Günnemann 2023a],要么通过任意旋转来增加训练数据 [Scherbela et al. 2023]。除了欧几里得群,势能面还对核的排列群 𝑆𝑀 具有不变性。通常通过在轨道函数中依赖核的求和而不是连接来实现这种对称性 [Gao and Günnemann 2021]。
多种几何形态 — 大小一致性:与对称性类似,大小一致性是有关系统能量的先验信息。具体而言,大小一致性是指能量随所建模系统的大小而变化。在极限情况下,系统可以分解为两个非相互作用的子系统,整个系统的能量就是子系统能量的总和。尽管如此,由于它仅表述在非相互作用系统的极限情况下,它不能严格地被表述为对称性。尽管如此,通过大小一致性来限制神经波函数的功能形式可以减少潜在函数类别的搜索空间,并实现更好的泛化 [Gao and Günnemann 2023a]。可以证明,为了实现大小一致性,需要限制轨道函数 𝜙 具有衰减的感受野,以便在足够远的距离内粒子不会相互作用 [Gao and Günnemann 2023a]。这与广泛使用的 FermiNet 架构不兼容,后者强烈依赖于全局平均值。分子轨道网络(Moon)[Gao and Günnemann 2023a]通过在类似于 SchNet [Schütt et al. 2017b] 的消息传递方案中依赖衰减的空间滤波器来实现这一点。
多种几何形态 — 能量曲面:由于波函数直接与能量相关,因此对于每个结构,可以数值地解决 ∫ 𝜓𝜃 (𝒓, 𝑀)𝐻ˆ𝑀𝜓𝜃 (𝒓, 𝑀)𝑑𝒓 以获得相应的能量。但是,由于蒙特卡洛积分本质上是昂贵的过程,如果需要评估成千上万个状态,这将代价高昂。对于单个结构计算,最终能量通常被近似为在训练期间观察到的能量的平均值,例如最后 20% 的观察到的训练能量。然而,这不能转化为通常从某种连续分布中采样分子结构的广义情况 [Gao and Günnemann 2021]。因此,Gao 等人提出了 Potential learning from ab-initio Networks (PlaNet) [Gao et al. 2022] 来解决这个问题。在 PlaNet 中,将从单分子设置中平均训练能量的想法转化为 PES 建模,方法是在每个时间步骤内平均观察到的能量曲面。实际上,这意味着在每个时间步骤,都会将一个函数拟合到观察到的能量上。然后,在一阶泰勒近似中,通过平均函数的参数来进行时间平均。
3.3.4 数据集和基准。
与量子自旋系统相同(第3.2.5节),训练数据是根据神经波函数定义的分布进行采样的。因此,无需事先生成数据集。相反,数据是根据几何结构的原子坐标来定义的。另一方面,由于优化过程的可变性,精度是通过从样本估计的平均能量以及标准偏差来评估的,更低的能量表示更准确的结果。常见测试系统包括小或重的原子,如N或Fe,小或大的分子,如N2或CCl4 [von Glehn et al. 2023],一些特殊的原子配置,如H10,化合物结构,如苯二聚体 [Ren et al. 2023; von Glehn et al. 2023],以及分子系统的跃迁能,定义为化学过程发生前后的基态能量差,例如环丁二烯(C4H4)的自动重排和Fe的电离 [Spencer et al. 2020]。
3.3.5 开放性研究方向。
使用VMC模拟多电子系统仍然存在一些挑战。首先,由于费米子反对称性约束,大多数现有方法使用Slater行列式。然而,通过行列式进行优化可能会引入额外的困难。目前尚不清楚我们是否可以使用除了Slater行列式之外的方法有效地实现费米子反对称性。其次,目前大多数方法都在实空间中显式地模拟波函数。在实现机器学习中的概率密度建模方面,朝着隐式建模的方向发展可能是一个有趣的方向。最后,最紧迫的挑战之一在于计算效率。由于计算复杂性随电子数量 𝑁 的增加呈 𝑂(𝑁^4) 的规模,当前的计算仅限于最多80个电子。进一步扩展这些方法非常重要。这可以通过更高效的采样、更好的优化以及在系统之间实现更有效的权重共享来实现。例如,深度学习库Jax [Bradbury et al. 2018] 已经通过改进实现在能量评估方面显示出了良好的加速性能。扩展是将QMC方法应用于更大的分子系统或材料的关键。