学习内容
-
应用似然比检验 (LRT) 进行假设检验 -
将 LRT 生成的结果与使用 Wald 检验获得的结果进行比较 -
从 LRT 显著基因列表中识别共享表达谱
似然比检验
在评估超过两个水平的表达变化时,DESeq2 还提供似然比检验作为替代方法。被确定为重要的基因是那些在不同因子水平上在任何方向上表达发生变化的基因。
通常,此测试将产生比单独的成对比较更多的基因。虽然 LRT 是对因子的任何水平差异的显着性检验,但不应期望它与使用 Wald 检验的基因集的并集完全相等(尽管我们确实期望高度重叠) 。
result
要从我们的 dds_lrt 对象中提取结果,我们可以使用与 Wald 检验相同的 results() 函数。不需要对比,因为我们没有进行成对比较。
# Extract results for LRT
res_LRT <- results(dds_lrt)
让我们看一下结果表:
# View results for LRT
res_LRT
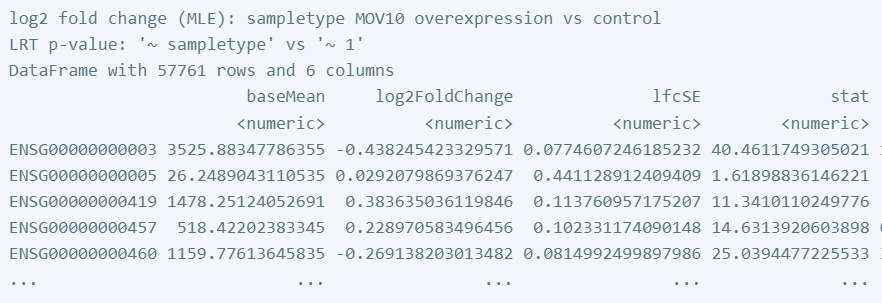
输出看起来类似于 Wald 检验的结果,具有与我们之前观察到的相同的列。
-
为什么要报告 LRT 检验的倍数变化?
对于使用似然比检验的分析,p 值仅由完整模型公式和简化模型公式之间的偏差差异决定。单个 log2 倍变化打印在结果表中以与其他结果表输出保持一致,但与实际测试无关。
与 LRT 检验相关的:
-
baseMean:所有样本的归一化计数的平均值 -
stat:简化模型和完整模型之间的偏差差异 -
pvalue:将统计值与卡方分布进行比较以生成pvalue -
padj:BH 调整后的 p 值
附加列:
-
log2FoldChange:log2 倍变化 -
lfcSE:标准错误
识别重要基因
当从 LRT 中过滤重要基因时,我们仅对 padj 列设置阈值。 padj < 0.05 时有多少基因是显著的?
# Create a tibble for LRT results
res_LRT_tb <- res_LRT %>%
data.frame() %>%
rownames_to_column(var="gene") %>%
as_tibble()
# Subset to return genes with padj < 0.05
sigLRT_genes <- res_LRT_tb %>%
filter(padj < padj.cutoff)
# Get number of significant genes
nrow(sigLRT_genes)
# Compare to numbers we had from Wald test
nrow(sigOE)
nrow(sigKD)
从 LRT 观察到的重要基因数量相当多。该列表包括可以在三个因子水平(控制、KO、过表达)中以任何方向变化的基因。为了减少重要基因的数量,我们可以增加 FDR 阈值 (padj.cutoff) 的严格性。
-
识别具有共享表达谱的基因簇
我们现在有了这份约 7K 重要基因的列表,我们知道这些基因在三个不同的样本组中以某种方式发生了变化。我们接下来做什么?
下一步是识别在样本组(水平)之间共享表达变化模式的基因组。为此,我们将使用来自“DEGreport”包的名为 degPatterns 的聚类工具。 degPatterns 工具使用基于基因间成对相关性的层次聚类方法,然后切割层次树以生成具有相似表达谱的基因组。该工具以优化集群多样性的方式切割树,使得集群间的可变性 > 集群内的可变性。
在我们开始聚类之前,我们将首先对我们的 rlog 转换归一化计数进行子集化,以仅保留差异表达的基因 (padj < 0.05)。在我们的例子中,对 7K 基因运行聚类可能需要一些时间,因此出于类演示目的,我们将子集化以仅保留按 p 调整值排序的前 1000 个基因。
# Subset results for faster cluster finding (for classroom demo purposes)
clustering_sig_genes <- sigLRT_genes %>%
arrange(padj) %>%
head(n=1000)
# Obtain rlog values for those significant genes
cluster_rlog <- rld_mat[clustering_sig_genes$gene, ]
重要基因的 rlog 转换计数与一些附加参数一起输入到 degPatterns:
-
metadata:样本对应的元数据dataframe -
time:元数据中的字符列名称,将用作更改的变量 -
col:元数据中的字符列名,用于分隔样本
# Use the `degPatterns` function from the 'DEGreport' package to show gene clusters across sample groups
clusters <- degPatterns(cluster_rlog, metadata = meta, time = "sampletype", col=NULL)
聚类运行完成后,您将在控制台中返回命令提示符,您应该会在绘图窗口中看到一个图形。这些基因被分为四个不同的组。对于每组基因,我们都有一个箱线图来说明不同样本组之间的表达变化。叠加了一个折线图来说明表达变化的趋势。
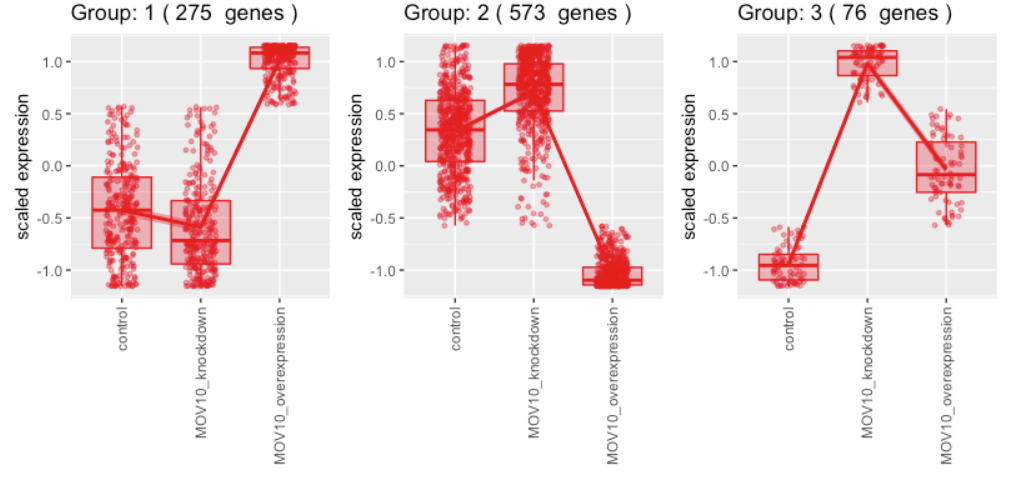
假设我们对在样本中表现出表达减少和过表达增加的基因感兴趣。根据该图,共有 275 个基因共享此表达谱。为了找出这些基因是什么,让我们探索一下输出。聚类输出的数据结构是什么类型?
# What type of data structure is the `clusters` output?
class(clusters)
我们可以使用名称(簇)查看列表中存储了哪些对象。里面存储了一个数据框。这是主要结果,让我们看一下。第一列包含基因,第二列包含它们所属的簇编号。
# Let's see what is stored in the `df` component
head(clusters$df)
由于我们对第 1 组感兴趣,我们可以过滤数据框以仅保留那些基因:
# Extract the Group 1 genes
group1 <- clusters$df %>%
filter(cluster == 1)
提取一组基因后,我们可以使用注释包来获取额外的信息。我们还可以使用这些基因列表作为下游功能分析工具的输入,以获得更多的生物学见解,并查看基因组是否共享特定功能。
欢迎Star -> 学习目录
更多教程 -> 学习目录
本文由 mdnice 多平台发布



![[阶段4 企业开发进阶] 7. 微服务](https://img-blog.csdnimg.cn/a353a99d6b8649b6a11c795c34a4f3d9.png#pic_center)