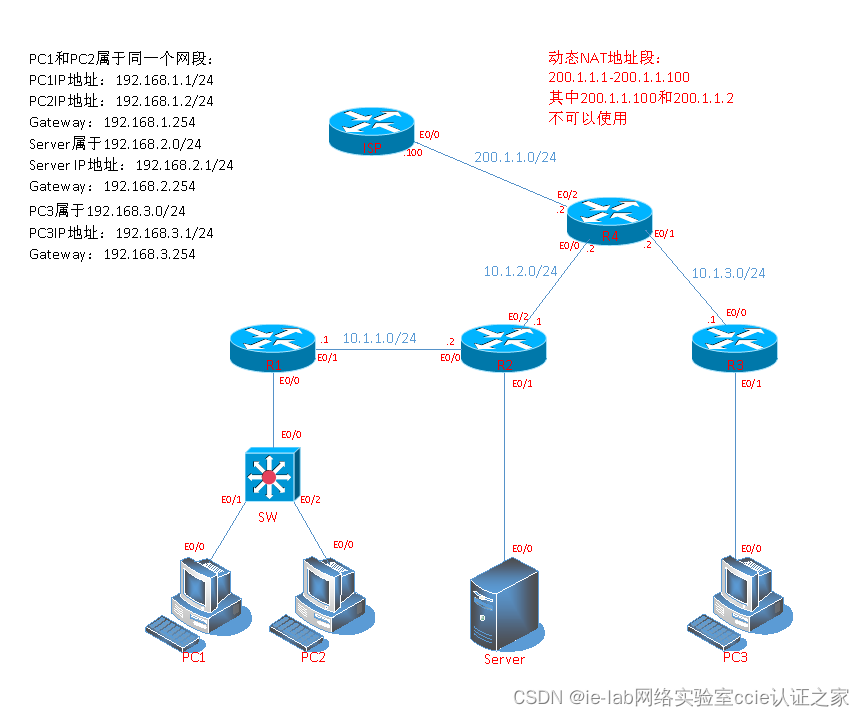
书接上文组学知识速递(五)|ChIP-seq知多少?,当我们实验完成,拿到下机数据之后,我们最关心的就是,这个数据能不能用?所谓数据能不能用,其实我们会重点关注以下问题:
1)fastq的测序质量过不过关?
2)实验本身有没有问题,处理组与对照组是否有区别?
3)分析结果是否能挖掘出有用或者新的信息?
接下来,一起来找寻答案吧!
Q1 ChIP-seq的分析一般有哪些步骤呢?
ChIP-Seq即染色质免疫共沉淀-高通量测序,是指通过染色质免
1)FastQC用于简单的质量控制(quality control),FastQ_Screen用于检查测序数据有无污染;
2)经过质控的reads通过bowtie2与参考基因组比对;
3)ChIP-seq peaks则用MACS2进行分析;
4)这些peaks则通过ChIPseeker进行注释,motif预测则使用HOMER;
5)最后Peak差异则使用MAnrom1。
Q2 有效数据量达到多少比较合适?
一般情况下,分析得到差异显著的峰的个数随着reads数目的增加而以稳定的比例增加(图中实线所示),这种情况下reads的数目没有饱和。但是,当对Chip样品和Input DNA样品的峰之间的差异定义一个最小的富集阈值后,分析得到的新峰的比率逐渐减小(图中虚线所示),这时,当分析足够具有显著差异peaks数目的时候,结合位点数目的饱和点出现,可以通过定义几个不同的阈值,分析几个曲线到达平台期的数值来定义饱和的标准(图中桔黄色线所示),所指定的阈值即为最小饱和富集比率(the minimum saturation enrichment ratio,MSER),所得到的最小饱和富集比率可以作为测序深度选择的参数。
当然一般的Human或者mouse的ChIP-seq数据选择20 million的数据就已经足够了。测序量不够,一些比较弱的信号可能就会被噪音给盖住。

Q3 比对率达到多少是合格的?
一般来说,Illumina 测序的样品比例应该超过80%。不过也有例外,像IgG这样的非dna结合蛋白的标记率通常较低(约60%)。当然,这些数字也不是绝对的,不是说80%可以,79%就不成,我们得根据实验设计来做具体判断。
80%以上的数据比对到了基因组上,说明至少样本没有出问题。至于数据能不能用,还得看peak calling步骤结果,或者可以用IGV大致看看有没有信号。
Q4 如何理解覆盖度累积曲线中反映的信号富集程度?
对样本比对结果reads累积情况进行展示。一定长度窗口(bin)上reads数进行计数,然后排序,再依次累加画图。input 在基因组上理论是均匀分布,随着测序深度增加趋近于直线,实验组在排序越高的窗口处reads累积速度越快,说明这些区域富集的越特异。
narrow peak :富集程度高;broad peak:富集程度低。富集程度低不代表失败, 如broad peak。但是如果是转录因子, 富集程度低则需要谨慎对待。

Q5 什么样的igv可视化图可表征特异性片段富集?

Q6 不同的组蛋白组结合区域有什么区别?
虽然大多数ChIP-seq工具都是针对特定基因组区域的sharp peaks,如转录起始位点(TSS),但一些组蛋白修饰与大基因组结构域相关,从而导致富集区域广泛分布。H3K27me3和H3K36me3富集分布在几百个碱基上,而H3K9me3 peaks通常扩展到几兆碱基。增强子标记H3K27ac和H3K4me1产生sharp peaks,但有时也会构建broad富集区域,称为“超级增强子”。H3K4me3启动子标记还可以覆盖小鼠卵母细胞中的broad结构域。这种peak形状和宽度变化影响最佳计算工具的选择。比如,ROSE用于检测超级增强子位点,Music用于计算要研究样本平均的peaks宽度。
Q7 不同的组蛋白call peak的区别是什么呢?
对于不同组蛋白call peaks要根据在基因组结合的模式来判断是narrow 或者broad peaks,然后再判断用何种方法去把相应的peaks 鉴定出来。在得到peaks list以后要随机在peaks list选取几个peaks拿到UCSC上去check一下,看是否这些peaks足够准确。如不够sensitive则需要根据情况调整参数。
Q8 Call peaks的工具该如何选择?
ChIP-seq技术经过多年的发展,已经开发出了很多call peaks的工具,例如FindPeaks、MACS、PeakSeq、SISSRs等等,而且也都有大量发表的高水平文章引用这些工具,常用的是MACS。然而需要注意的是对ChIP-seq数据进行call peaks分析需要具体问题具体分析,这是由于不同的蛋白以及表观遗传学修饰在基因上分布的pattern是非常不一样的,有H3K4me3那样非常sharp的peaks,也有H3K27me3那样非常broad的peaks。因此针对不同的ChIP-seq应该用不同的工具。一般针对于peaks比较sharp的ChIP-seq 数据用MACS14,而针对peaks比较 broad的ChIP-seq数据,用MACS2 callpeaks broad模式。
Q9 怎么知道结合的位置是broad还是sharp呢?用igv看吗,还是有什么评估的方法?
主要先用IGV或者UCSC genome browser先看一下ChIP-seq的pattern更像哪一种patttern,然后再决定使用哪种工具。
Q10 如何在ChIP-seq结果中寻找目标富集的Motif?
有些蛋白是直接结合DNA,此种情况下,基于peak的motif预测结果,查找是否有自己的目标蛋白;
有些蛋白是与其它蛋白互作,间接结合在DNA上,此种情况下,建议先查下自己的目标蛋白是否有互作蛋白,然后再基于peak的motif预测结果,查找motif list中是否有与自己的目标蛋白互作的蛋白。