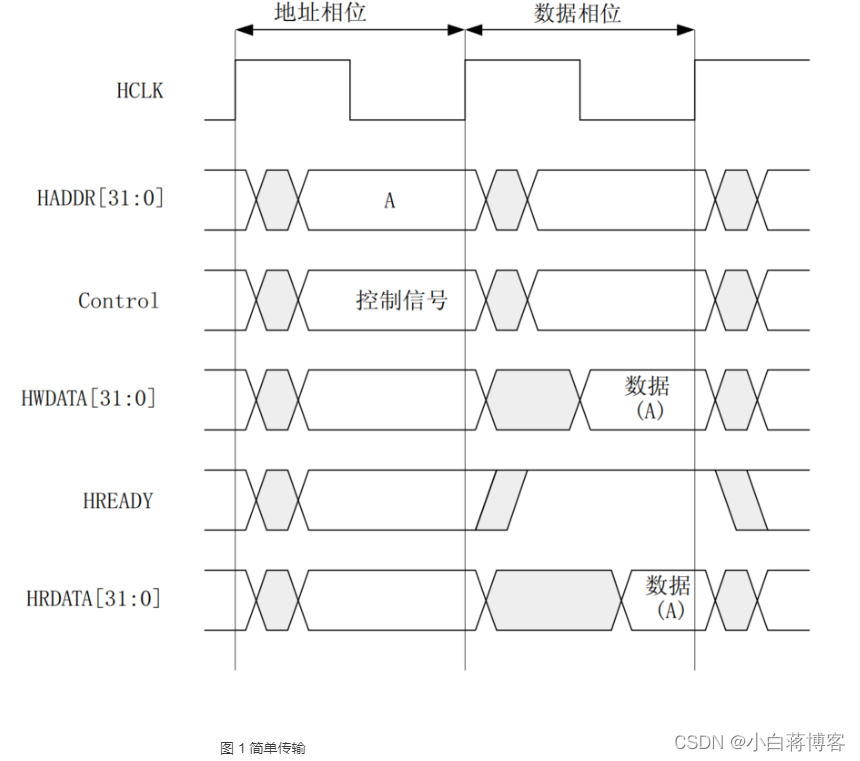
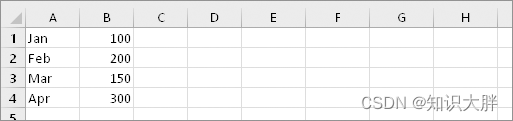
主成分回归(PCR)的方法 本质上是使用第一个方法的普通最小二乘(OLS)拟合来自预测变量的主成分(PC)。这带来许多优点:
- 预测变量的数量实际上没有限制。
- 相关的预测变量不会破坏回归拟合。
但是,在许多情况下,执行类似于PCA的分解要明智得多。
最近我们被客户要求撰写关于PLS-DA的研究报告,包括一些图形和统计输出。
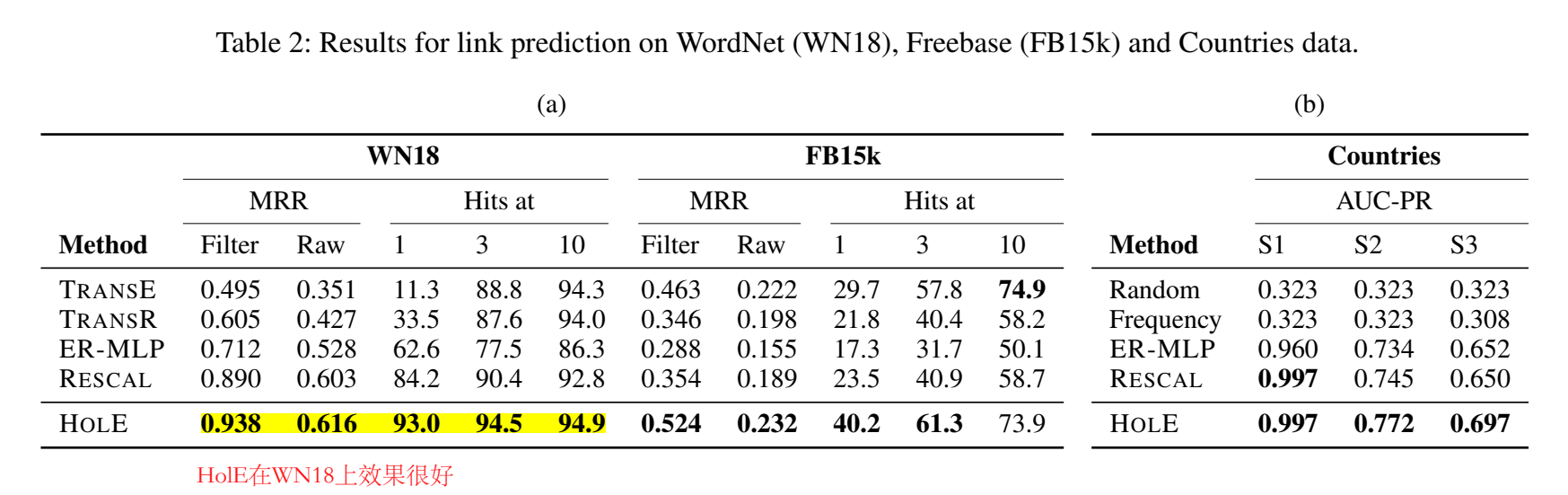
主成分分析PCA降维方法和R语言分析葡萄酒可视化实例
主成分分析PCA降维方法和R语言分析葡萄酒可视化实例
,时长04:30
今天,我们将 在Arcene数据集上执行PLS-DA, 其中包含100个观察值和10,000个解释变量。
让我们开始使用R
癌症/无癌标签(编码为-1 / 1)存储在不同的文件中,因此我们可以将其直接附加到完整的数据集,然后使用公式语法来训练模型。
# 安装加载
library(caret)
arcene <- read.table("train.data", sep = " ",
colClasses = c(rep("numeric", 10000), "NULL"))
# 将标签添加为附加列
arcene$class <- factor(scan("rain.labels", sep = "\t"))现在的主要问题是:
- 我们如何根据其血清的MS谱准确预测患者是否生病?
- 哪种蛋白质/ MS峰最能区分患者和健康患者?
关于预处理,我们将使用preProc参数以精确的顺序删除零方差预测变量,并对所有剩余的变量进行标准化。考虑样本的大小(n= 100),我将选择10次重复的5折交叉验证(CV)–大量重复弥补了因减少的验证次数而产生的高方差–总共进行了50次准确性估算。
# 编译交叉验证设置
set.seed(100)
myfolds <- createMultiFolds(arcene$class, k = 5, times = 10)
control <- trainControl("repeatedcv", index = myfolds, selectionFunction = "oneSE")
此图描绘了CV曲线,在这里我们可以学习从使用不同数量的LV(x轴)训练的模型中获得的平均准确度(y轴,%)。
现在,我们 进行线性判别分析(LDA)进行比较。 我们还可以尝试一些更复杂的模型,例如随机森林(RF)。
最后,我们可以比较PLS-DA,PCA-DA和RF的准确性。
我们将使用resamples编译这三个模型,并借用ggplot2的绘图功能来比较三种情况下最佳交叉验证模型的50个准确性估计值。
显然,长时间的RF运行并没有转化为出色的性能,恰恰相反。尽管三个模型的平均性能相似,但RF的精度差异要大得多,如果我们要寻找一个鲁棒的模型,这当然是一个问题。在这种情况下,PLS-DA和PCA-DA表现出最好的性能(准确度为63-95%),并且这两种模型在诊断新血清样品中的癌症方面都表现出色。
总而言之,我们将使用PLS-DA和PCA-DA中预测的变量重要性(ViP)确定十种最能诊断癌症的蛋白质。
上面的PLS-DA ViP图清楚地将V1184与所有其他蛋白质区分开。这可能是一个有趣的癌症生物标志物。当然,必须进行许多其他测试和模型来提供可靠的诊断工具。





![[附源码]Python计算机毕业设计Django疫情网课管理系统](https://img-blog.csdnimg.cn/f6acbf77028e4469a443516d34e8f943.png)

![[附源码]JAVA毕业设计体育用品购物系统(系统+LW)](https://img-blog.csdnimg.cn/54dfaf2e9d384752bb5b06b8f930db0a.png)