https://www.cnblogs.com/yanzhi123/p/11712926.html![]() https://www.cnblogs.com/yanzhi123/p/11712926.html【3】蛋白质组学鉴定软件之Mascot - 简书 (jianshu.com)
https://www.cnblogs.com/yanzhi123/p/11712926.html【3】蛋白质组学鉴定软件之Mascot - 简书 (jianshu.com)
【6】蛋白质组学鉴定定量软件之MaxQuant - 简书 (jianshu.com)
基于Maxquant软件处理的LabelFree蛋白质组学
首先,在使用Maxquant软件进行查库的时候,有两个参数值得大家关注:LFQ和iBAQ。
当我们在进行搜库时,如果两个参数都选择,将会在结果文件中有三个定量结果:Intensity,IBAQ和LFQ。
接下来,我们来详细解释一下这三个定量结果的区别:
Intensity:将Protein Group中的所有Unique和Razor peptides的信号强度求和,作为最原始的强度值
iBAQ:基于 Intensity 的强度值,除以该蛋白的理论肽段数目,主要用于同一样本内,不同蛋白的相互比较
LFQ:基于 Intensity 的强度值,在不同样本间执行矫正操作,达到降低样本预处理,仪器分析等操作造成的样本间的差异,主要用于同一蛋白,不同样本间的表达差异。比如,不同处理方法间,同一蛋白的表达量变化
【盘点】代谢组学中常5大分析方法 - 质谱 - 实验与分析
蛋白质组学基础入门系列 |(一) 蛋白质组学的基本概念 - 简书
蛋白质组学基础入门系列 |(二)蛋白质组学研究利器——质谱 - 简书
Mascot search engine | Protein identification software for mass spec data (matrixscience.com)
蛋白质组学基础入门系列 |(三)蛋白质组学常用技术盘点(上) - 简书
非标记定量技术
基于标记的蛋白质组学定量技术
蛋白质组学基础入门系列 |(四)蛋白质组学常用技术盘点(下) - 简书
蛋白质组学基础入门系列丨(五)蛋白质组学样品制备流程 - 简书
蛋白质组学基础入门系列丨(六)蛋白质组学质谱检测环节 - 简书
蛋白质组学基础入门系列丨(七)蛋白质组学数据库检索 - 简书
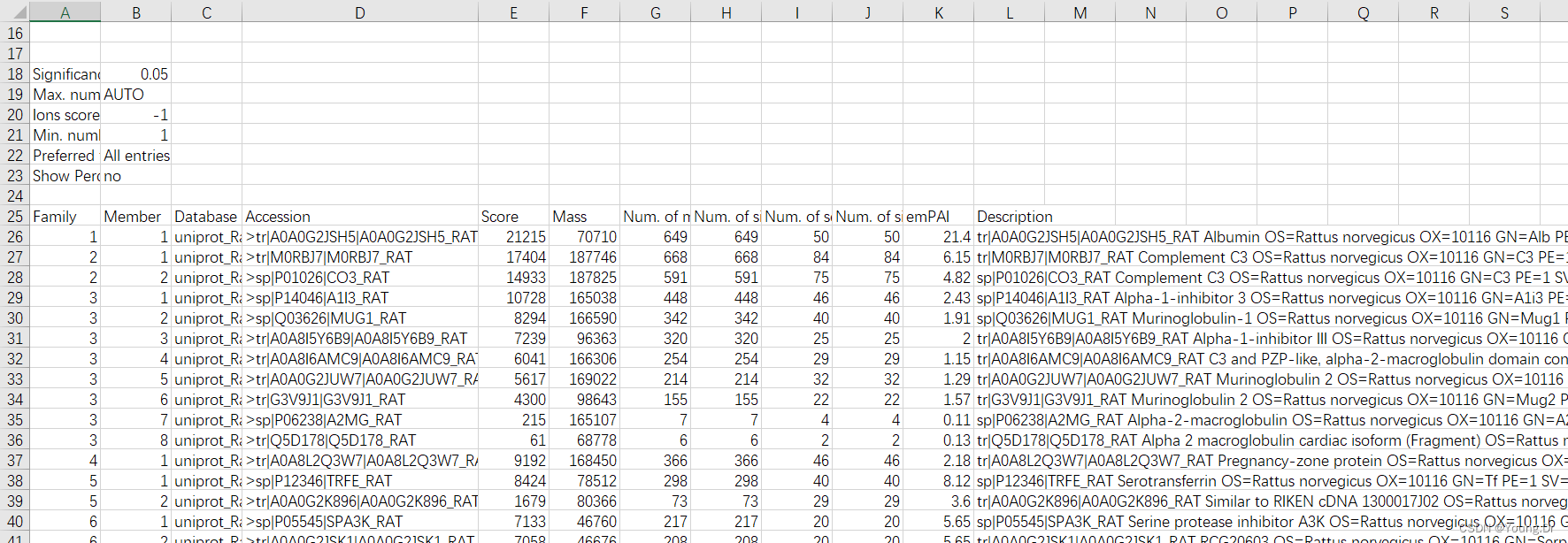
In the context of protein sequence identifiers, "sp" and "tr" are prefixes used to indicate different databases or data subsets.
- "sp": The "sp" prefix stands for Swiss-Prot, which is a high-quality manually curated protein sequence database maintained by the Universal Protein Resource (UniProt). Swiss-Prot provides comprehensive and reliable protein sequence information, including functional annotations, post-translational modifications, and protein-protein interactions. The identifiers starting with "sp" indicate protein sequences from the Swiss-Prot database.
- "tr": The "tr" prefix stands for TrEMBL (Translated EMBL Nucleotide Sequence Data Library), which is a computer-annotated protein sequence database also maintained by UniProt. TrEMBL contains protein sequences that are automatically generated from the translation of coding sequences from the EMBL-Bank nucleotide sequence database. The identifiers starting with "tr" indicate protein sequences from the TrEMBL database.
Both Swiss-Prot and TrEMBL are parts of the UniProt database, but Swiss-Prot contains manually curated and reviewed entries, while TrEMBL consists of computationally annotated entries. The prefix "sp" indicates proteins from Swiss-Prot that have undergone more extensive curation, while the prefix "tr" indicates proteins from TrEMBL that may have less curated information.
-
"tr": The prefix "tr" stands for "TrEMBL." It refers to protein entries in the UniProtKB/TrEMBL subsection of the UniProt Knowledgebase. These entries are computationally predicted and automatically annotated using various algorithms and tools. TrEMBL entries contain protein sequences and annotations that have not yet undergone manual curation and review. They serve as a comprehensive and rapidly growing collection of protein data, providing a resource for researchers to access a wide range of protein sequences and functional predictions. The "tr" prefix indicates that the protein entry is from the unreviewed section of UniProtKB.
-
"con": The prefix "con" is used to denote protein sequences that are artificial or synthetic constructs. These protein sequences may be artificially designed or engineered for experimental purposes, such as studying protein structure-function relationships or investigating specific protein domains. The "con" prefix indicates that the protein entry represents a synthetic or artificial protein construct.
蛋白组学定量值得比较说明
1. Maxquant的iBAQ和LFQ,该用哪个?
我们使用Maxquant做Label Free蛋白质组学定量分析的时候,在Maxquant的参数设置时,会遇到两个参数,LFQ和iBAQ,那么,选择哪个好呢?
如果你都选上,在最终的proteingroups.txt中,会出现三列:Intensity、IBAQ、LFQ intensity,这三列中的数字,也就是蛋白的定量强度,并不一样,那么,到底那一列比较准呢?
首先,让我们来看一下三者的计算原理是什么?
> Intensity是将某Protein Groups里面的所有Unique和Razor peptides的信号强度加起来,作为一个原始强度值。
> iBAQ是在上面的基础上,将原始强度值除以本蛋白的理论肽段数目。
> LFQ则是将原始强度值在样本之间进行校正,以消除处理、上样、预分、仪器等造成的样本间误差。
假设有两个蛋白,A和B,A和B在样本中的量是相等的,也就是等量。 假设A的长度是10个肽段,B的是100个肽段,假设鉴定结果中,覆盖度都是30%,那么蛋白A的强度是3,B的是30,。这时候我们对比一下,B是A的10倍,但是,A和B原本是相等,这样就存在较为严重的误差。
这时候,如果我们将其原始强度值除以理论肽段数目,A的强度变成了3/10, B的强度变成了3/10。 A = B,Perfect!
上面就是IBAQ的原理和用处。
但是在定量蛋白质组学中,我们并不做蛋白A和 B之间的定量,假如你有一个药物处理前的细胞和药物处理后的细胞的对照型样本做的定量蛋白质组学实验,我们关注的蛋白A在处理前和处理后的变化,至于A和B之间的比值,并不重要。
所以,如果是样本内对比,当然用iBAQ,因为其表征的是蛋白的摩尔比值(copy number)。如果是样本间对比,当然是LFQ(正式名称为MaxLFQ,也就是搜库结果中的txt文件中的LFQ Intensity)[1]
当然,如果你执意要用iBAQ,你可以手工校准样本件误差,方法很简单:蛋白IBAQ值除以此样品所有蛋白的强度的和,计算比例(这也是组学中“等质量上样”和“等体积上样”的核心区别,等质量上样来看的是比例,但是计算比例是有压缩效应的)[2]。
最后,总结一下:
同一个(或者说同一针)样品内部的蛋白互相比较,用IBAQ;
不同样品间互相比较(不管是重复还是不同的处理组),用LFQ。
Reference:
[1]Cox J, Hein M Y,Luber C A, et al. Accurate Proteome-wide Label-free Quantification by DelayedNormalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ[J]. Molecular& Cellular Proteomics Mcp, 2014, 13(9):2513.
[2]Shin J B, Krey JF, Hassan A, et al. Molecular architecture of the chick vestibular hairbundle[J]. Nature Neuroscience, 2013, 16(3):365-74.
Significance A and B for protein ratios
实验设计中,一般会做三个生物学重复来确保结果的准确性,尤其在下游分析中。但有时会遇到没有生物学重复,而又需要进行差异分析的情况,这时一般建议考虑foldchange即可,因为根本无法进行T-test等统计学方法嘛。但是如果必须要算一个P值(个人觉得没啥必要。。。),那么不同组学有各自处理的方法(虽然并不是靠谱),比如NGS的转录组的一些软件会预估一个离散度做校正,而质谱的蛋白组则是用Significance A/B算法,这篇文章主要讲下Significance A/B是怎么来的
一般在网上搜Significance A/B是很难搜到相关信息的,因为这个是特定用于蛋白组学的一种统计学方法,而且现在来说用的也比较少了;那当初为何提出这分析方法,个人觉得可能是因为那时蛋白组学成本过高。以前一直只知道有这一分析方法,但是不知其原理,最近在搜索中无意发现一个帖子What statistical methods for ITRAQ with two biological replication?,其中提到一篇文章中有对Significance A/B的介绍
Significance A/B最先是发表于2008年Nature Biotechnology期刊上,MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification,这篇文章主要是介绍Maxquant这款用于蛋白组定量分析软件的,非常有名,而其附录中作者提到了如何通过protein ratio来计算显著性(P值)
代码实现
了解了上述的Significance A/B的计算过程,那么我们就可以用代码将其实现,下面我用R写了个函数来计算Significance A,而Significance B从上述可知,只要对protein分bin后再用Significance A计算即可(这里不重复展示了),输入为ratio向量
get_significance <- function(ratio){
ratio <- log2(as.numeric(ratio))
order_ratio <- ratio[order(ratio)]
quantiletmp <- quantile(order_ratio, c(0.1587,0.5,0.8413))
rl <- as.numeric(quantiletmp[1]) #对应公式中的r-1
rm <- as.numeric(quantiletmp[2]) #对应公式中的r0
rh <- as.numeric(quantiletmp[3]) #对应公式中的r1
p <- unlist(lapply(ratio, function(x){
if (x > rm){
z <- (x-rm)/(rh-rm)
pnorm(z,lower.tail = F)
}else{
z <- (rm-x)/(rm-rl)
pnorm(z,lower.tail = F)
}
}))
}
p <- get_significance(data)
http://www.bioinfo-scrounger.com Introduction to proteomics data analysis: MaxLFQ Summarization![]() https://statomics.github.io/SGA2020/assets/cptac_maxLfQ.html
https://statomics.github.io/SGA2020/assets/cptac_maxLfQ.html