梳理一下scanpy单细胞分析流程(处理的是scRNA-seq)。
先上一张流程图:
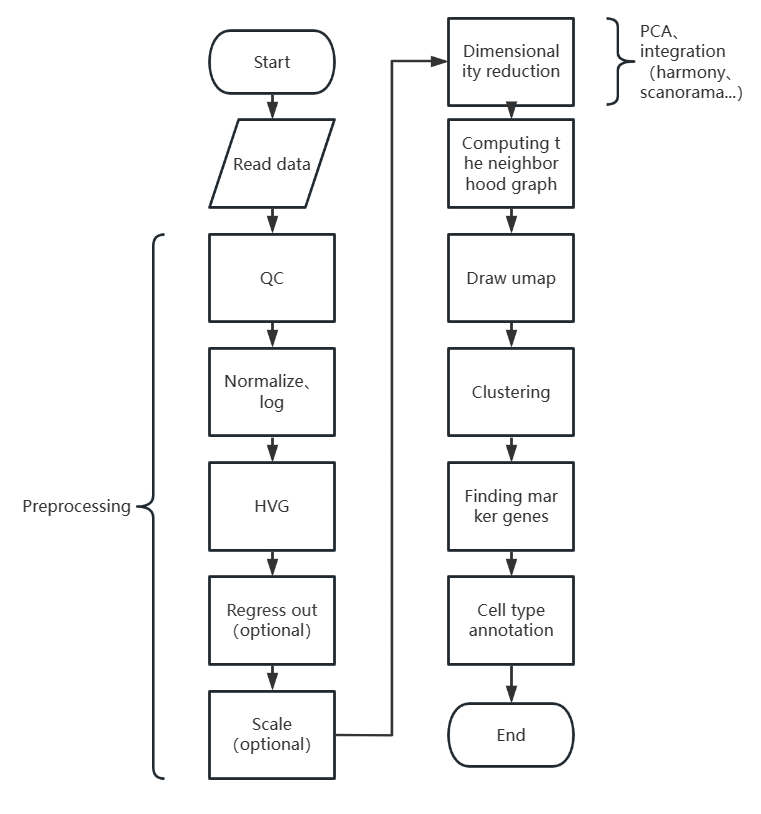
scanpy单细胞分析流程
import scanpy as sc
Read data
常用的文件格式有两种,分别是h5ad和10X mtx
# read h5ad
adata = sc.read()
# read 10X mtx
adata = read_10x_mtx()
Preprocessing
QC
QC这一步目标是过滤掉低质量的细胞和基因
我们通常可以通过以下指标进行QC
- Cell counts
- UMI counts per cell
- Genes detected per cell
- Complexity(novelty score)
- Mitochondrial counts ratio
Cell counts
这个指标可以与预期的细胞数进行对比,用来判断是否有垃圾细胞存在。需要注意的是,
UMI counts per cell
UMI(unique molecular identifiers),用来给mRNA计数
The UMI counts per cell should generally be above 500, that is the low end of what we expect. If UMI counts are between 500-1000 counts, it is usable but the cells probably should have been sequenced more deeply.
Genes detected per cell
每个细胞表达的基因数,在scanpy的教程中,过滤掉了低于200的细胞
novelty score
什么是novelty score
We can evaluate each cell in terms of how complex the RNA species are by using a measure called the novelty score. The novelty score is computed by taking the ratio of nGenes over nUMI. If there are many captured transcripts (high nUMI) and a low number of genes detected in a cell, this likely means that you only captured a low number of genes and simply sequenced transcripts from those lower number of genes over and over again. These low complexity (low novelty) cells could represent a specific cell type (i.e. red blood cells which lack a typical transcriptome), or could be due to an artifact or contamination. Generally, we expect the novelty score to be above 0.80 for good quality cells.
novelty score公式
This value is quite easy to calculate, as we take the log10 of the number of genes detected per cell and the log10 of the number of UMIs per cell, then divide the log10 number of genes by the log10 number of UMIs. The novelty score and how it relates to complexity of the RNA species, is described in more detail later in this lesson.
Mitochondrial counts ratio
This metric can identify whether there is a large amount of mitochondrial contamination from dead or dying cells. We define poor quality samples for mitochondrial counts as cells which surpass the 0.2 mitochondrial ratio mark, unless of course you are expecting this in your sample.
在scanpy中,这一值被设定为5%
合理的值范围可能是5%—20%
Joint filtering effects
Considering any of these QC metrics in isolation can lead to misinterpretation of cellular signals. For example, cells with a comparatively high fraction of mitochondrial counts may be involved in respiratory processes and may be cells that you would like to keep. Likewise, other metrics can have other biological interpretations. A general rule of thumb when performing QC is to set thresholds for individual metrics to be as permissive as possible, and always consider the joint effects of these metrics. In this way, you reduce the risk of filtering out any viable cell populations.
Two metrics that are often evaluated together are the number of UMIs and the number of genes detected per cell. Here, we have plotted the number of genes versus the number of UMIs coloured by the fraction of mitochondrial reads. Jointly visualizing the count and gene thresholds and additionally overlaying the mitochondrial fraction, gives a summarized persepective of the quality per cell.
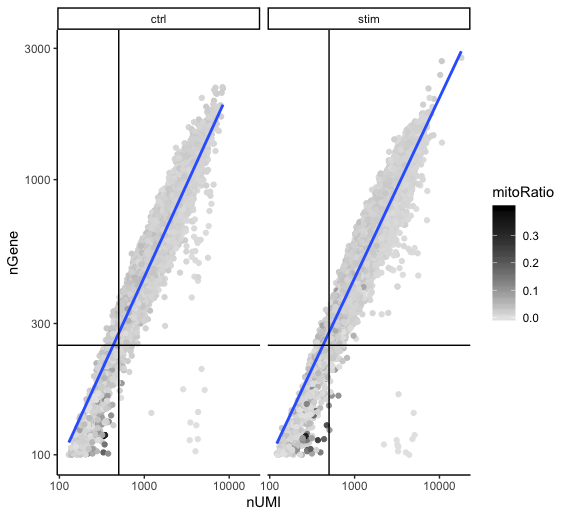
Good cells will generally exhibit both higher number of genes per cell and higher numbers of UMIs (upper right quadrant of the plot). Cells that are poor quality are likely to have low genes and UMIs per cell, and correspond to the data points in the bottom left quadrant of the plot. With this plot we also evaluate the slope of the line, and any scatter of data points in the bottom right hand quadrant of the plot. These cells have a high number of UMIs but only a few number of genes. These could be dying cells, but also could represent a population of a low complexity celltype (i.e red blood cells).
Normalize、log
Normalize
Normalize each cell by total counts over all genes, so that every cell
has the same total count after normalization.
为啥要Normalize?
normalize让细胞的rna-seq具有组件可比性,也就是让细胞之间可以对比
log
Logarithmize the data matrix.
为啥要log?
取log之后可以方便计算,同时在找差异表达基因时需要log后的数据
更多的统计学解释可以参考这个问题下的讨论:在统计学中为什么要对变量取对数?
HVG
筛选出细胞之间表达量差别大的基因,方便下游对比
scanpy实现了三种算法
Seurat, Cell Ranger and Seurat v3
不同算法需要的条件可能不同
看详情,点这里
Regress out
Regress out (mostly) unwanted sources of variation.
并不是必要的,可能会过度矫正
Scale
Scale data to unit variance and zero mean.
也就是z-score normalization,别称Standardization
不是必要的
Dimensionality reduction
将数据降维,方便下游的近邻图计算,常用方法是PCA。
integration
在这一步中,我们也可以使用其他方法来代替PCA,这些方法多是使用算法求得整个表达矩阵的embedding,以这些embedding来代替表示表达矩阵,从而达到batch remove的效果。
常用的算法有scvi、harmony、scanorama等,更多的算法对比可以看scib这篇文章,这边文章做了一个benchmark的工作,将现有的主流integration算法进行了对比评估。
康康我的这篇博文里有文章地址和常用评估代码。
单细胞数据integration结果评估
Computing the neighborhood graph
Compute a neighborhood graph of observations.
这一步计算出的近邻图是下面绘制umap和聚类需要用到的。
Draw umap
绘制umap图
Clustering
聚类,将细胞分成簇。常用的有louvain和leiden两种算法。常用的是leiden。
Find marker gene
求某一簇相对于其他簇的差异表达基因
Cell type annotation
细胞类型标注,这一步有两种方法,一种是基于marker gene对细胞进行标注,一种是利用机器学习的方法对细胞进行自动标注,如single r。
scanpy教程链接
Preprocessing and clustering 3k PBMCs



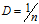








![[PyTorch][chapter 43][时间序列表示方法1]](https://img-blog.csdnimg.cn/6203bc9d3ade432c91fc63c5e3768157.png)