ExamPle: explainable deep learning framework for the prediction of plant small secreted peptides
期刊:Bioinformatics
影响因子:6.931
中科院分区:小类数学与计算生物2区
出版日期:2023年3月10日
Github:https://github.com/Johnsunnn/ExamPle
摘要
植物小分泌肽(SSPs)在植物生长发育和植物与微生物相互作用中起着重要作用。在过去的几十年里,基于机器学习的方法得到了发展,然而,现有方法高度依赖手工特征工程,容易忽略潜在的特征表示,影响预测性能。
在这里,我们提出了一个新的深度学习模型,使用Siamese网络(Siamese network)和多视图表示(multi-view representation)来预测植物ssps的可解释性。基准比较结果表明,我们的示例在植物ssps预测方面的性能明显优于现有方法。同时,我们的模型也表现出了出色的特征提取能力。重要的是,通过利用硅诱变(silico mutagenesis)实验,ExamPle可以发现序列特征并确定每个氨基酸对预测的贡献。我们的模型学到的关键新原理是肽的头部区域和一些特定的序列模式与ssps的功能密切相关。因此,该方法有望成为预测植物ssps和设计有效植物ssps的有效工具。
数据集
植物SSPs数据集:我们从MtSSPdb (Boschiero et al . 2020)中收集植物ssps数据集的阳性样本。MtSSPdb是一个综合性的数据库,包含在模式豆科植物紫花苜蓿中发现的植物ssp。在阴性样本中,我们发现了两个非分泌肽家族CYSTM和DVL (Butenko et al . 2009;Xu et al . 2018);之后,我们搜索非ssp序列并从Pfam数据库下载,该数据库是一个蛋白质家族的大型集合(Mistry et al 2021)。之后,我们使用CD-Hit (Li和Godzik, 2006)将数据集中序列的相似性降低到20%。此外,正负样本平衡的数据集通常有利于深度学习模型的训练过程。因此,我们从两个数据库中获取相同数量的阳性和阴性样本。按照上述步骤,我们得到了植物ssp数据集,包括1184个植物分泌肽和1184个植物非分泌肽。将数据集按80%和20%的比例分别划分为训练和测试数据集。
肽二级结构:在本研究中,我们使用PHAT web界面生成肽二级结构。PHAT由Jiang等人(2023)提出。PHAT是一种用于预测肽二级结构的新型深度学习框架。PHAT利用强大的预训练蛋白语言模型和一种新型的超图多头注意网络,不仅可以将语义知识从大尺度蛋白质转移到多肽,学习多肽残基的高潜伏和长期特征,还可以对多肽残基进行多语义二级结构信息编码,同时利用多层次注意机制捕获连续区域的上下文特征。此外,PHAT具有交互式、无代码和非编程的web界面。我们使用PHAT web界面获取预测的肽的3态二级结构,然后将3态二级结构信息添加到植物ssp数据集中。
方法
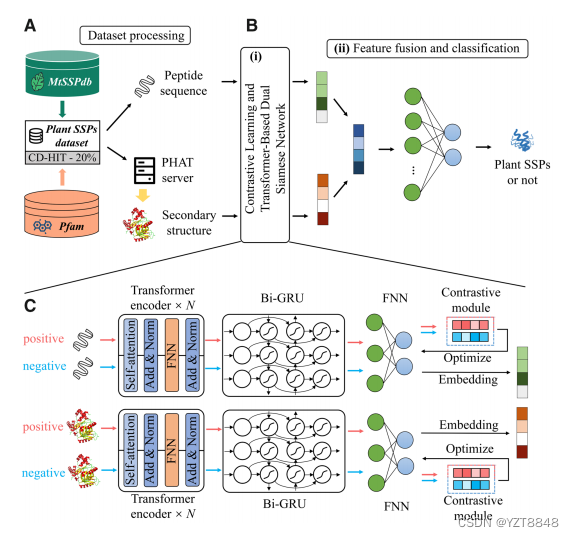
框架概述
本文提出了一种基于两个Transformer和两个Bi-GRU结构和Siamese网络的预测框架,该框架融合了多肽序列和二级结构信息来预测植物ssps。我们的框架架构如图1所示,包括B(i)对比学习和基于变压器的双暹罗网络和B(ii)特征融合和分类。
首先,在B(i)中,我们将植物ssp序列及其二级结构输入到基于双变压器的Siamese网络模型中进行特征嵌入。具体来说,每个基于Transformer的Siamese网络包括Transformer编码器、Bi-GRU(双向门循环单元)、全连接神经网络(FNN)和对比模块。两个Transformer模型不共享权重。变压器编码器用于生成ssp序列和二级结构的上下文敏感特征表示。然后,利用Bi-GRU捕获具有长距离信息的特征表示。随后,使用FNN嵌入特征以生成统一的表示。
然后,我们可以得到两个ssp特征表示之间的距离。对比模块计算对比损失,利用对比损失减小相同标签的表示向量之间的距离,增大不同标签的表示向量之间的距离。其次,在B(ii)中,我们融合了从B(i)中学习到的序列和二级结构特征。随后,我们使用FNN作为鉴别器。根据鉴别器给出的预测分数,该模型可以判断给定肽是否属于植物ssp。如果预测分数大于0.5,则属于植物ssp。
Transformer
Transformer由Vaswani等人(2017)提出,最初应用于机器翻译任务,并已成为大量自然语言处理(NLP)任务中最先进的模型。与之前基于蛋白质能量的方法相比(Mrozek et al 2007;2009;Malysiak-Mrozek and Mrozek 2011), AlphaFold通过使用Transformer (Jumper et al 2021)取得了巨大的成功。因此,在这项工作中,我们采用Transformer作为模型的主干。在模型的Transformer部分,我们使用Transformer的原始版本。变压器由编码模块和解码模块组成。编码模块由一系列编码器组成,用于提取输入的特征。此外,解码模块由一系列用于生成表示的解码器组成。在这里,我们只使用Transformer编码器提取特征。变压器编码器层如图所示
GRU
门控循环单元(GRU)由Cho等人(2014)提出。它旨在使每个循环单元自适应地捕获不同时间尺度的依赖关系。与长短期记忆(LSTM)相比,GRU的架构得到了简化(Hochreiter and Schmidhuber 1997),同时保持了与LSTM相同的有效性。GRU具有门控单元,可以像LSTM单元一样调节单元内的信息流,但没有单独的存储单元。LSTM单元的三个基本组成部分——输入门、遗忘门和输出门在GRU中被转换成两个门:
Siamese network and contrastive module
暹罗网络可以用来量化两个序列的相似性。例如,为了寻找匹配和不匹配的图像对,Melekhov等人(2016)使用Siamese网络架构中的对比损失函数,利用卷积神经网络从匹配和不匹配图像对的标记示例中学习得到特征向量。Li和Zhang(2019)提出了一种名为SiamVGG的连体网络,该网络结合了卷积神经网络骨干和相互关联算子,利用示例图像的特征进行更精确的目标跟踪。具体地说,给定两个输入向量v1和v2,它们被输入到相同的模块中。网络将输入映射到相同的特征空间。然后,网络使用距离测量函数计算两个输入的相似度,记为d - v1;v2Þ。这里,我们使用欧几里得距离d / v1;v2Þ:
结果
ExamPle with other methods
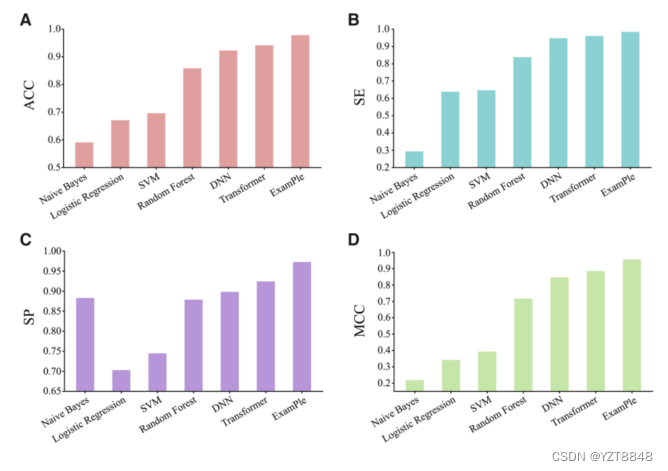
为了评估ExamPle的性能,我们采用了几种经典的比较模型,包括朴素贝叶斯(Rish 2001)、逻辑回归(Hosmer et al . 2013)、支持向量机(SVM) (Cortes and Vapnik 1995)、随机森林(Breiman 2001)、深度神经网络(DNN) (Montavon et al . 2018)和标准Transformer,考虑到之前的工作中缺乏直接预测ssp的方法。为了公平比较,我们使用训练集进行模型训练,并在相同设置的独立数据集中评估这些模型。为了更好地再现对比实验,DNN和Transformer的架构由DeepBIO (Wang et al 2022)服务器实现。如图2所示,ExamPle在ACC、SE、SP和MCC指标方面优于所有比较模型。例如,ExamPle的ACC性能比Transformer提高了3.65%,比Random Forest提高了11.99%;MCC的性能比DNN提高了10.91%,比SVM提高了56.35%。总之,这些结果表明,ExamPle有效预测植物ssp的能力优于大多数机器学习和深度学习方法。
ExamPle特征与其他特征的比较
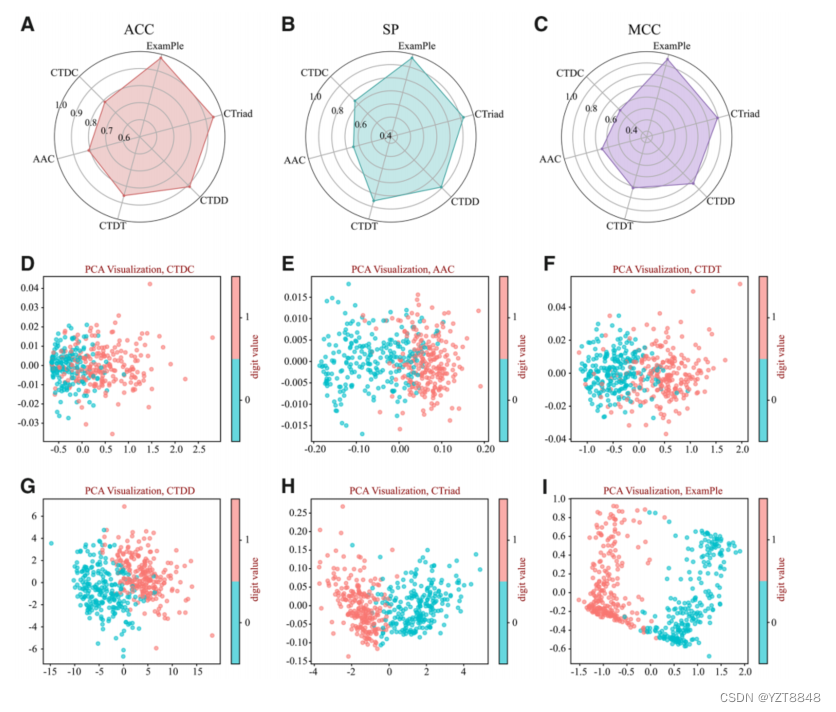
为了验证通过示例学习的特征表示的有效性,我们将不同的手工特征方法与我们的模型进行了性能比较。在iLearnPlus (Chen et al . 2018,2020,2021)的帮助下,我们选择了5种特征编码方法,包括氨基酸组成(AAC) (Spackman et al . 1958)、CTD组成(CTDC)、CTD分布(CTDD)、CTD过渡(CTDT) (Dubchak et al .)1999)和联合三位一体(CTriad) (Shen et al . 2007)。这些手工特征编码的详细信息可在补充表S1中找到。然后,我们分别将五种手工特征编码方法输入到模型的分类层中进行训练和测试。如图3所示,CTriad编码方法在ACC上达到94.67%,CTDC编码方法达到78.86%。然而,由ExamPle嵌入的表示优于所有其他手工制作的特征编码方法。特别是在MCC性能方面,我们的模型比CTDC高37.57%,比CTDT高24.28%。为了进一步验证示例的性能,我们可视化了五种手工特征编码方法嵌入的特征表示以及使用降维的模型。如图3(D-I)所示,我们可以更直观地看到,我们的模型编码的表征比其他任何五种手工制作的特征表征都能更好地分类。综上所述,与其他手工特征编码方法相比,我们的模型具有良好的适应性和鲁棒性,可以学习到更强大的特征表示。
可视化
在训练过程中,深度学习模型是如何工作的尚不清楚。为了更透明地理解,我们使用主成分分析(Abdi and Williams 2010)和t分布随机邻居嵌入(t-SNE) (Van der Maaten and Hinton 2008)将模型的潜在空间减少到二维空间,以验证ExamPle的效率。如图4所示,在训练过程开始时,阴性样本和阳性样本混合在一起。随后,随着训练迭代次数的增加,样本逐渐分为两组,表明我们的模型通过反向传播优化可以学习到足够的信息。

分析
鉴于我们的复杂神经网络架构的优异性能,我们的目标是从计算的角度进一步探索植物ssp的序列特征。为此,我们对数据集中的每个肽进行了硅诱变(ISM)解释方法,并将一个肽的每个参考氨基酸突变为其他19个替代氨基酸,以评估预测的概率变化。此外,我们在图5A和B中分别对ssp数据集和非ssp数据集上的每个位置进行了氨基酸统计概率,作为我们分析的参考。
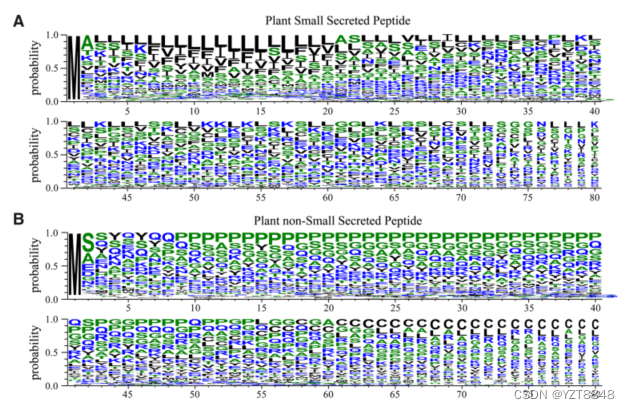
在这里,我们选择一个59-bp的序列作为样本,生成如图5C所示的热图,其中正值表示该序列被某个氨基酸取代后预测得分较高,负值表示预测得分较低。这张热图很容易显示出氨基酸Q(谷氨酰胺)和P(脯氨酸)对预测分数有显著的抑制作用。具体来说,在10 ~ 16位,当氨基酸发生Q或P突变时,预测分数显著降低。这可能是因为Q和P在非ssp中频繁出现,而在ssp中出现的频率较低,如图5A和b所示。因此,当识别序列中的Q和P时,模型倾向于将序列视为非ssp。特别是,C (l -半胱氨酸)经常出现在非ssp的尾部。图5C的观察结果表明,氨基酸C显著降低了尾部区域的预测分数。此外,氨基酸I (L -异亮氨酸)、L(亮氨酸)和F(苯丙氨酸)在序列的大部分位置对预测分数有促进作用。可能的原因是,如图5A所示,I、L、F在ssp序列中出现频率较高,而在非ssp序列中出现频率较低。在识别序列中的I、L和F时,模型倾向于将序列视为ssp。

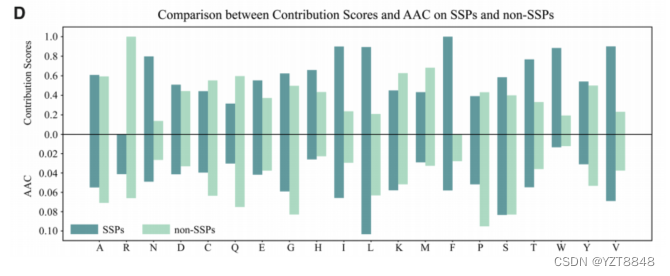
然后,为了使结果更具统计意义,我们计算了每个氨基酸对整个数据集的贡献。对于ssp和非ssp的每个肽,我们通过累积ISM结果中的每个位置评分来计算氨基酸评分。具体来说,我们可以将图5C中一行的得分相加,得到20个值,每个值代表这个59- bp长度序列中20个氨基酸的贡献得分。根据每个肽的氨基酸贡献得分,我们将它们相加,分别估算出对SSP和非SSP的综合氨基酸贡献得分。
不同氨基酸在ssp数据集和非ssp数据集中的贡献结果如图5D所示。此外,我们利用氨基酸组成(AAC)作为统计分析参考。从图5D可以看出,对于绝大多数氨基酸,每种氨基酸在ssp或非ssp中对预测评分的相对贡献与该氨基酸在ssp或非ssp中的相对AAC是一致的。简而言之,对于特定的氨基酸,如果它在ssp数据集中的AAC高于非ssp,则对预测分数的贡献可能在ssp中高于非ssp。此外,我们可以看到ssp中氨基酸G(甘氨酸)的AAC小于非ssp,但我们的模型认为G在ssp中对预测得分的贡献更大,表明我们的模型可以挖掘传统统计方法无法计算的特征。综上所述,这些实验表明我们的模型可以挖掘出ssp的序列特征。
功能与序列区域之间的联系
接下来,我们试图了解功能影响与序列区域之间的联系。为此,无论一个位置的积极或消极影响,我们使用从ISM实验中减去的绝对值作为重要性分数来评估上下文区域如何影响植物ssp的功能。为了将所有的序列统一成一个固定的长度,我们将序列分成50个部分。在这里,每个部分被称为一个位置比率。

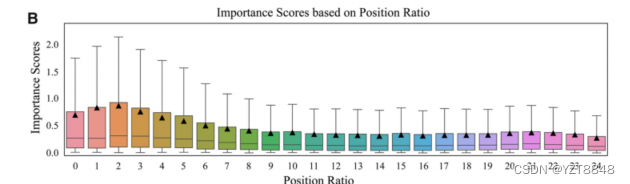
在图6A中,我们给出了整个数据集中氨基酸与位置比的完整相关图。颜色越深,重要性得分越高。例如,氨基酸M(蛋氨酸)对中心区域的功能影响更大,而氨基酸C(半胱氨酸)对尾部区域的功能影响更大,这都是肽设计中值得注意的有意义的点。接下来,为了进一步直观地说明哪个区域更重要,我们将数据集中的每个肽分成25个部分,并累积氨基酸得分,得到该位置的综合得分。从图6B中可以明显看出,头部区域的重要分数呈上升趋势,中部区域的重要分数呈缓慢下降趋势。在位置比率1和3之间,重要性得分的中位数是位置比率9后面的中位数的两倍。

为了更好地验证图6B中的现象,我们进行了一个裁剪实验,分别从正向和反向对序列进行裁剪,并将裁剪后的序列输入到模型中,考察裁剪后的序列是否能保持原有的功能。从图6C可以看出,后向削波性能非常稳健,在削波比增加到0.5之前,精度一直保持在0.9以上。然而,一旦裁剪操作开始,前向裁剪的度量迅速下降,当裁剪比超过0.7时,整个数据集的ACC甚至低于0.5。有趣的是,衡量阴性情况准确性的度量SP在裁剪开始时保持相同的水平,这足以证明阴性序列不依赖于头部区域,而可能与中心区域相关。

在我们利用重要性分数直观地找到哪个区域对肽函数是重要的之后,从计算的角度来探索这个区域的基序是很自然的。在图6D中,我们根据重要分数发现了一个基本基序,并将其与传统工具streme搜索的基序进行了比较(Bailey 2021)。可以看到,我们学到的母题(用灰色窗口突出显示)与STREME的母题非常相似。然后,我们利用TOMTOM (Bailey et al . 2009)对Pvalue测量的基序相似度进行定量计算。p值越低,基序一致性越高。图6D显示,我们选择的基序与STREME的基序高度相似,表明我们的模型可以发现保守的序列模式。综上所述,这些实验结果表明,我们的模型ExamPle有潜力成为序列模式发现的新工具,并可能为稳定有效的植物ssp设计铺平道路。
结论
在这项研究中,我们提出了一个新的可解释的对比混合视图双暹罗框架例子,用于预测植物ssp和发现ssp的序列模式。实验结果表明,具有二级结构信息的Siamese框架能够学习到判别特征信息,有效地提高了模型对植物ssp预测的性能。此外,通过采用降维方法,潜在空间的可视化清晰地显示了我们的模型具有很强的学习能力,优于传统的手工特征工程。利用ISM解释方法,ExamPle可以发现序列特征,确定各氨基酸的贡献函数,与统计结果基本一致。此外,通过积累变异序列和原始序列之间的功能差异,我们的模型学到的关键新原理是肽中的头部区域和一些特定的序列模式与SSP功能密切相关。因此,我们训练良好的模型ExamPle不仅可以提供植物ssp的预测,而且可以为有效和稳定的植物ssp设计铺平道路。







![[冷冻电镜]IMOD使用指南](https://img-blog.csdnimg.cn/3beb3da0651a4797ae9bc5e26f4b4c8e.png)