欢迎关注我的CSDN:https://spike.blog.csdn.net/
本文地址:https://blog.csdn.net/caroline_wendy/article/details/131095395
PDB 三大数据集的多维度分析与整理:
- 人工提交 - RCSB PDB:PDB Database - RCSB PDB 数据集的多维度分析与整理 (1)
- 算法预测 - AlphaFold DB:PDB Database - AlphaFold DB PDB 数据集的多维度分析与整理 (2)
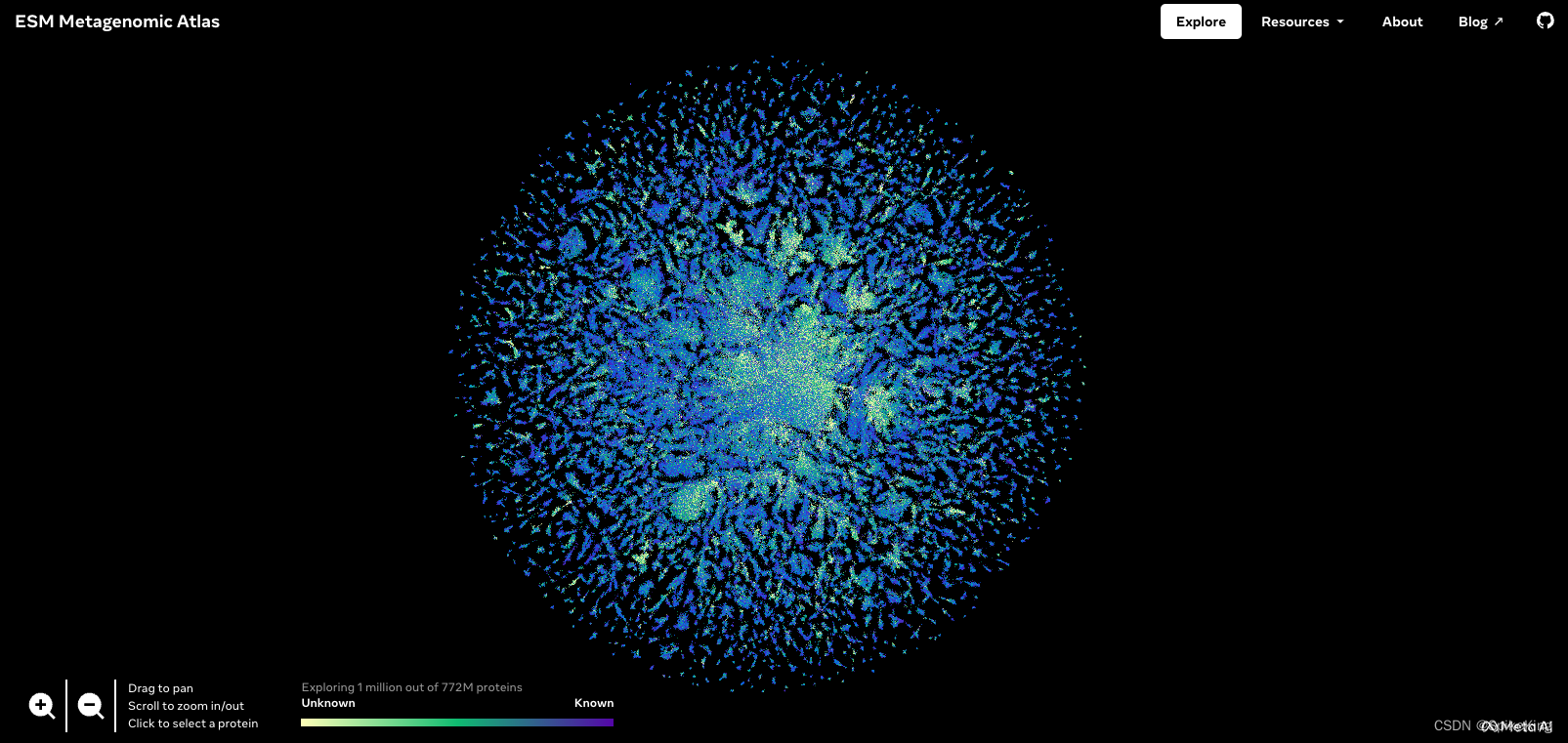
- 算法预测 - ESM Metagenomic Atlas:PDB Database - ESM Atlas PDB 数据集的多维度分析与整理 (3)
ESM Metagenomic Atlas 数据集是一个包含了来自不同环境样本的微生物基因组序列的大型数据库。该数据集的目的是为了探索微生物的多样性和功能,以及它们与环境因素的相互作用,使用了高通量测序技术,对来自全球各地的土壤、水、空气等样本进行了深入的分析,该数据集为微生物生态学和环境科学提供了一个宝贵的资源。
1. ESM Metagenomic Atlas 介绍
官网:https://esmatlas.com/
数据网站:https://github.com/facebookresearch/esm/tree/main/scripts/atlas
- 数据规模1.08T
The ESM Metagenomic Atlas is a repository of over database of more than 600 million metagenomic protein structures predicted by ESMFold. We are releasing the 600+ million protein ESM Metagenomic Atlas, with predictions for nearly the entire MGnify90 database, a public resource cataloging metagenomic sequences.
- ESM 宏基因组图谱是一个超过 6 亿个由 ESMFold 预测的宏基因组蛋白质结构的数据库。我们正在发布 600+ 百万蛋白质 ESM 宏基因组图谱,其中包含几乎整个 MGnify90 数据库的预测,这是一个宏基因组序列的公共资源目录。
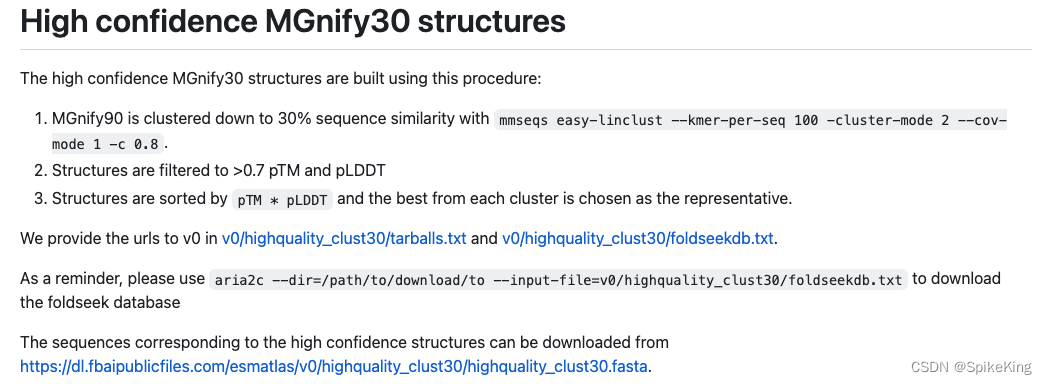
2. High Confidence MGnify30 介绍
重要的是高置信度部分,即 High confidence MGnify30 structures,高置信度 MGnify30 结构:
The high confidence MGnify30 structures are built using this procedure:
- MGnify90 is clustered down to 30% sequence similarity with
mmseqs easy-linclust --kmer-per-seq 100 -cluster-mode 2 --cov-mode 1 -c 0.8.- MGnify90 聚类到 30% 的序列相似性
- Structures are filtered to >0.7 pTM and pLDDT.
- Structures are sorted by
pTM * pLDDTand the best from each cluster is chosen as the representative.
其中,FASTA文件:highquality_clust30.fasta,大约7.5G
>MGYP000000000040
MCGVYQSATFQATFFQYSYILHETLADIVVPDTIGGKIRKLRHSLNLAQMQFAKSIHRGFTTVTKWEQELTTTSEKALTNIIEIYKLQENYFDK
>MGYP000000000300
MSNTPDDKNFDIAGFLLAGNIMVKLVEKGVIDMRDANDIVARTRAAYTQRDSYKDESLGSDAEAYLDTLFNKLWASRPDAVGKK
>MGYP000000000306
MKESKIIAMELSIKALNEDFAYFRFGEHFSGFIHKTGNGCATIILDGGYLLGVYESADEALKGIAALGANILMAELKAGVSFSTYKSAYWPKNHNVH
>MGYP000000000315
MSLEMFQNQVRSTITELILKQSALFGAATGGAMMLGSEKTIGDYAEESSWKLIAGLVTARNAYSTAPVTAKEIEQILKRAPRFDWRMGPAKVNDGLLARINSSPEDAAAAISAQASQGIIEQQITQGLAALDACLSTNEKFSLAIAADSTAETGEITPKLSSFVKGRRAFGDAGQNIICWAINSDVYYSLVENDLFKNAEQLYKLGDISVFTDGLNGRFLVTDYVPSNTAYGMVNGAVIIDNGYQSKFVAQPQLGGESLTTMMQSEGEFKIAVKSYRIKSTLADTLANTVSITADQVKDPDNWEYVASDESRAQPGVKLTFTPKA
>MGYP000000000576
MSCSTVTVVRPRVWIGCLACYNEGRLVGEWVDAADAGDLTPDDLHGVPTTHEELWVFDLEGFPRGTGEMSPTASVPWGELFEEVGEAQWPALLAWVESGCYMADADGLPCVSDFEDAYQGEWDSFDDYAVQLAEDIGLTDGWPEDTQRYFNWDSWTRDLAFDYAVADASDGGVFVFRSC
默认聚类命令,参考 GitHub - MMseqs2: ultra fast and sensitive sequence search and clustering suite (超快速和灵敏的序列搜索和聚类套件):
mmseqs easy-linclust --kmer-per-seq 100 --cluster-mode 2 --cov-mode 1 -c 0.8
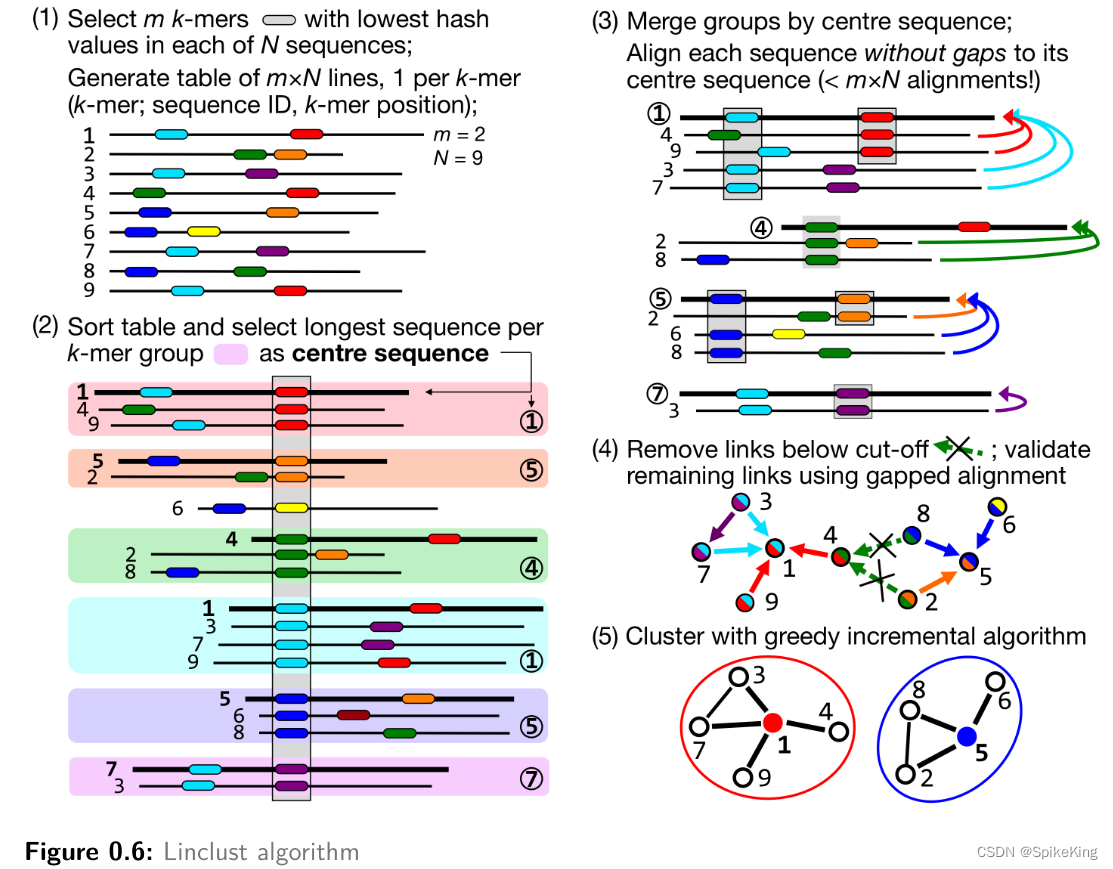
easy-linclust:- Linclust is a clustering in linear time. It is magnitudes faster but a bit less sensitive than clustering.
- Linclust 是一种线性时间聚类,比聚类快几个数量级,但是敏感度稍低一些。
-
--kmer-per-seq 100:- Increasing the k-mers selected per sequence increases the sensitivity of linclust at a moderate loss of speed. Use the parameter --kmer-per-seq to set the number of k-mers selected per sequence. More k-mers per sequences results in a higher sensitivity.
- 增加每个序列选择的k-mer数量,可以提高linclust的灵敏度,但是稍微降低速度。 使用参数
–kmer-per-seq来设定每个序列选择的 k-mer 数量。 每个序列的 k-mer 数量越多,灵敏度越高。
-
--cluster-mode 2:-
The second clustering algorithm is a greedy clustering algorithm (–cluster-mode 2), as used in CD-HIT. It sorts sequences by length and in each step forms a cluster containing the longest sequence and sequences that it matches. Then, these sequences are removed and the next cluster is chosen from the remaining sequences.
-
第二种聚类算法是一种贪婪聚类算法(
--cluster-mode 2),与CD-HIT中使用的相同。 按长度对序列进行排序,并且在每一步形成一个聚类,包含最长序列和匹配序列。 然后,这些序列被移除,从剩余的序列中,选择下一个聚类。
-
--cov-mode 1:- MMseqs2 has three modes to control the sequence length overlap “coverage”: --cov-mode (0) bidirectional, (1) target coverage, (2) query coverage and (3) target-in-query length coverage.
- MMseqs2有三种模式来控制序列长度重叠“覆盖率”:
--cov-mode(0) 双向,(1) 目标覆盖率,(2) 查询覆盖率 和 (3) 目标在查询长度覆盖率。
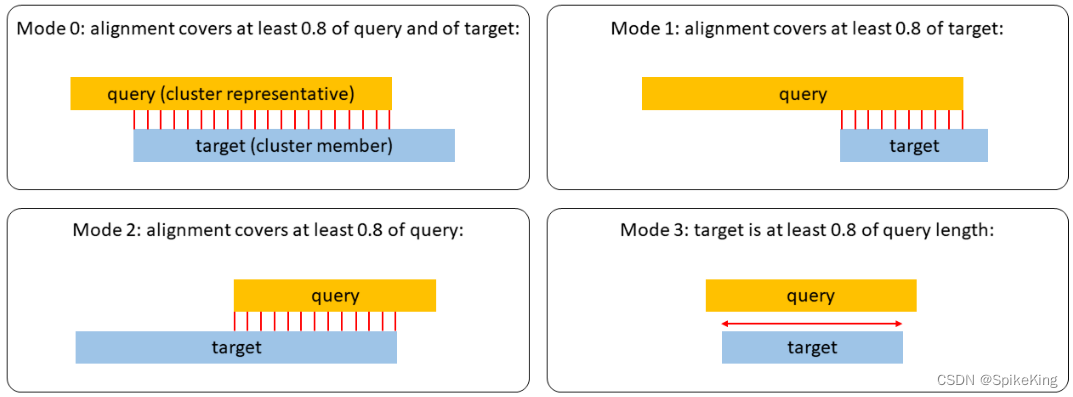
-c 0.8:- With --cov-mode 1 -c [0.0,1.0] (target-cov mode) only sequences are clustered that have a sequence length overlap greater than X% of the target sequence. The target cov mode can be used to cluster protein fragments. To suppress fragments from becoming representative sequences, it is recommended to use --cluster-mode 2 in conjunction with --cov-mode 1. Default --cluster-mode is the greedy incremental clustering (by length).
- 使用
--cov-mode 1 -c [0.0,1.0](目标覆盖率模式) 只有序列长度重叠大于目标序列的 X% 的序列才会被聚类。目标覆盖率模式,可用于聚类蛋白质片段。为了抑制片段成为代表性序列,建议使用 --cluster-mode 2 与 --cov-mode 1 结合使用。默认的 --cluster-mode 是贪婪增量聚类 (按长度)。
其他重要参数:
-
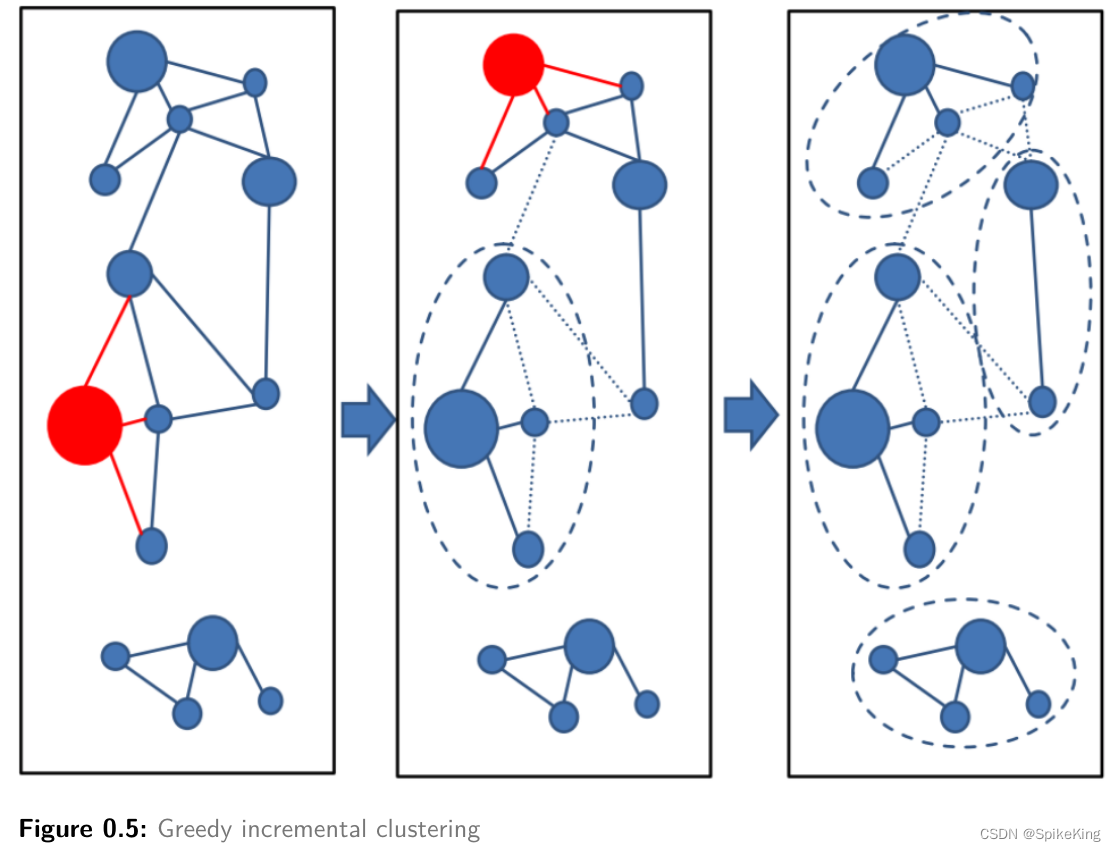
--min-seq-id 0.9:- MMseqs2/Linclust and Linclust has three main criteria, inferred by a local alignment, to link two sequences by an edge:
- MMseqs2/Linclust 和 Linclust 有三个主要标准,通过局部比对推断,将两个序列通过一条边连接起来:
- (3) a minimum sequence identity (–min-seq-id [0,1]) with option --alignment-mode 3 defined as the number of identical aligned residues divided by the number of aligned columns including internal gap columns, or, by default, defined by a highly correlated measure, the equivalent similarity score of the local alignment (including gap penalties) divided by the maximum of the lengths of the two locally aligned sequence segments. The score per residue equivalent to a certain sequence identity is obtained by a linear regression using thousands of local alignments as training set.
- (3) 最小序列一致性 (
--min-seq-id [0,1]),选项--alignment-mode 3定义成 相同对齐残基数量 除以 对齐列数(包括内部间隔的列),或者在默认情况下 ,由高度相关度量定义,局部比对的等效相似性得分(包括空位罚分) 除以 两个局部比对序列片段长度的最大值。使用数千个局部比对作为训练集,通过线性回归,获得相当于某个序列同一性的每个残基的分数。
-
--db-load-mode 2:- The touchdb module fetches the precomputed index database into memory and --db-load-mode 2 tells MMseqs2 to mmap the database instead of copying the whole precomputed index into memory. This saves, for a large database, minutes of copying from the storage system into RAM. However, this is less efficient for large query sets.
- 模块 touchdb 将预先计算的索引数据库提取到内存中,
--db-load-mode 2告诉 MMseqs2 映射数据库,而不是将整个预先计算的索引复制到内存中。 对于大型数据库,这可以节省从存储系统复制到 RAM 的时间。 然而,这对于大型查询集来说效率较低。
-
mmseqs easy-linclust,三个参数,输入fasta,输出clusterRes,临时文件夹tmp
K-mer:
k-mer是指生物序列中长度为k的子串。通常,k-mer是指一个序列的所有长度为k的子序列,例如,序列AGAT有四个单体(A,G,A和T),三个2-mer(AG,GA,AT),两个3-mer(AGA和GAT)和一个4-mer(AGAT)。在计算基因组学和序列分析的背景下,k-mer主要由核苷酸(即A,T,G和C)组成,用于组装DNA序列12、改善异源基因表达123、在宏基因组样本中鉴定物种1 以及创建减毒疫苗1 。在蛋白质序列中,k-mer由氨基酸组成,可以用于表征蛋白质的特征和构建系统发育树。
MSI (minimum sequence identity):
在蛋白质序列中,最小序列相似度是指两个序列之间的相同氨基酸的百分比。最小序列相似度是用于评估两个序列是否具有同源性或结构相似性的一个重要指标。一般来说,最小序列相似度越高,两个序列之间的结构和功能的保守性越高。在蛋白质序列聚类或同源建模的过程中,最小序列相似度可以作为一个筛选标准,以减少冗余和提高准确性。不同的聚类或建模方法可能有不同的最小序列相似度阈值,但是通常认为30%以上的序列相似度是可靠的。
MMSeqs2简介
MMseqs2: ultra fast and sensitive sequence search and clustering suite. MMseqs2 (Many-against-Many sequence searching) is a software suite to search and cluster huge protein and nucleotide sequence sets. MMseqs2 is open source GPL-licensed software implemented in C++ for Linux, MacOS, and (as beta version, via cygwin) Windows. The software is designed to run on multiple cores and servers and exhibits very good scalability. MMseqs2 can run 10000 times faster than BLAST. At 100 times its speed it achieves almost the same sensitivity. It can perform profile searches with the same sensitivity as PSI-BLAST at over 400 times its speed.
超快速和灵敏的序列搜索和聚类套件。MMseqs2(多对多序列搜索)是一个软件套件,用于搜索和聚类巨量蛋白质和核苷酸序列集。MMseqs2 是使用 C++ 实现的开源 GPL 许可软件,适用于 Linux、MacOS 和(作为测试版,通过 cygwin)Windows。该软件被设计为在多个内核和服务器上运行,并表现出非常好的可扩展性。MMseqs2 的运行速度比 BLAST 快 10000 倍。在其速度的 100 倍时,可以达到几乎相同的灵敏度。可以超过其 400 倍的速度执行与 PSI-BLAST 相同灵敏度的配置文件搜索。PSI,Position-Specific Iterative,位置特定的迭代。
整体的数据集,有36个压缩包,大约30G左右,命名方式如下:
highquality_clust30_00.tar.gz
highquality_clust30_01.tar.gz
highquality_clust30_02.tar.gz
highquality_clust30_03.tar.gz
...
highquality_clust30_36.tar.gz
解压tar.gz文件,需要提前mkdir,解压命令如下,单个文件解压时间 约1天 左右:
mkdir highquality_clust30
tar -xvf highquality_clust30_[00-36].tar.gz -C highquality_clust30/
解压之后,包括数字编号文件夹,000 - 999,文件夹编号表示最后3位的数字,例如992文件夹中的PDB文件:
MGYP003624135992.pdb
MGYP003624171992.pdb
MGYP003624208992.pdb
MGYP003624265992.pdb
MGYP003635273992.pdb
解压全部的数据集,约3600万:
nohup tar -zxf highquality_clust30_01.tar.gz -C ./highquality_clust30/ &
nohup tar -zxf highquality_clust30_02.tar.gz -C ./highquality_clust30/ &
...
nohup tar -zxf highquality_clust30_36.tar.gz -C ./highquality_clust30/ &
查看全部的解压流程,预计1天左右:
ps -aux | grep "tar -zxf"
3. Highquality Clust 30 抽样分析
以压缩包 highquality_clust30_36.tar.gz 为例,分析ESMFold预测宏基因组结构。highquality_clust30_36,包括1000个文件夹,997338个样本,则36个大约997338*36=36000000,约3600万。发布时间是2022-11-01,参考:
The first
v0version of the Atlas was released on November 1st 2022, corresponding to the sequences in the2022_05release of the MGnify protein database described here.
提取PDB的信息,例如:
,pdb,plddt,release_date,seq,len
0,MGYP000001592000,0.9167,2022-07-24,MKLSEFILLSESEKKWLVTHRASPLAQRTYPHLIVFLFQLEDYYVEAYCNIADKKIDEYRVLPNTNAIRHYLEAIPIDG,79
1,MGYP000002113000,0.8722,2022-07-24,MRFLIIDADADYRQLLRYHLEVEWPDAAIDELQPNGALALPERVRLGDTDLVLLGHPLAHERGFEWLSLLRSRTDCPPVILFAAESDEFLAVDALKAGAANFFPKARVRHNRLIDAVRAELHVGL,125
2,MGYP000002905000,0.8233,2022-07-24,FYRDEWPALRARHPDRFRLRLLFSRSRGERVTVEEVRREMEGFLDPATSLAFVCGPNRPREAAGPDGVRRREPGFCDLWCGSARRKQEGLLARVGFSPDRIRTEMW,106
3,MGYP000008090000,0.8254,2022-07-24,MMTDSVSGSVTGAYAAGQGDRAARFGIGQLVRHVLFDFRGVVFDIDPQFSDTEEWLLAIPEAVRPEKDQPFYHLLAENGDICYVAYASEGNLCPDDTGMPLRHPQAELIFERFENGRYLLKSRLAN,126
注意:在数据集中,有一些PDB是没有任何结构信息,只有文件头部,例如 MGYP003442707068.pdb:
HEADER 18-OCT-22
TITLE ESMFOLD V0 PREDICTION FOR MGYP003442707068
REMARK 1
REMARK 1 REFERENCE 1
REMARK 1 AUTH ZEMING LIN, HALIL AKIN, ROSHAN RAO, BRIAN HIE, ZHONGKAI ZHU,
REMARK 1 AUTH 2 WENTING LU, NIKITA SMETANIN, ALLAN DOS SANTOS COSTA,
REMARK 1 AUTH 3 MARYAM FAZEL-ZARANDI, TOM SERCU, SALVATORE CANDIDO,
REMARK 1 AUTH 4 ALEXANDER RIVES
REMARK 1 TITL LANGUAGE MODELS OF PROTEIN SEQUENCES AT THE SCALE OF
REMARK 1 TITL 2 EVOLUTION ENABLE ACCURATE STRUCTURE PREDICTION
REMARK 1 REF
REMARK 1 REFN
REMARK 1 PMID
REMARK 1 DOI 10.1101/2022.07.20.500902
REMARK 1
REMARK 1 LICENSE AND DISCLAIMERS
REMARK 1 ESM METAGENOMIC STRUCTURE ATLAS DATA IS AVAILABLE UNDER
REMARK 1 A CC-BY-4.0 LICENSE FOR ACADEMIC AND COMMERCIAL USE.
REMARK 1 COPYRIGHT (C) META PLATFORMS, INC. ALL RIGHTS RESERVED.
REMARK 1 USE OF THE ESM METAGENOMIC STRUCTURE ATLAS DATA IS SUBJECT
REMARK 1 TO THE META OPEN SOURCE TERMS OF USE AND PRIVACY POLICY.
预处理 esm_metagenomic_atlas_pdb_36_997338_labels_997310.csv,由997338个PDB提取出997310个信息,空PDB是28,0.0028%。
具体信息,如下:
- PDB样本总数: 997310
- plddt range: 0.5519 ~ 0.9898
- plddt分布: 60: 30, 70: 122794, 80: 465557, 90: 408929, sum: 997310
- seq len range: 20 ~ 1279
- len > 20: 997310, len < 20: 0
- 序列长度: 0: 302067, 100: 381539, 200: 193863, 300: 67256, 400: 29153, 500: 11832, 600: 5662, 700: 2989, 800: 1574, 900: 704, 1000: 671
pLDDT的分布:
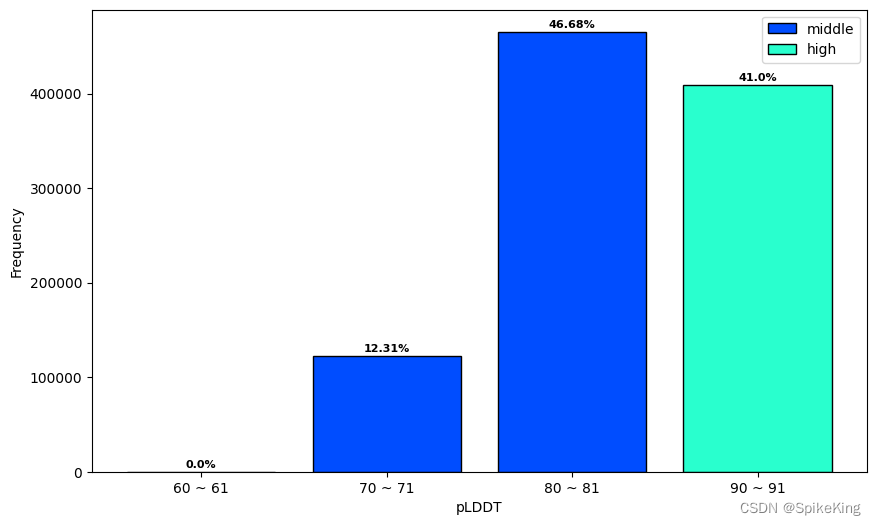
序列长度的分布:
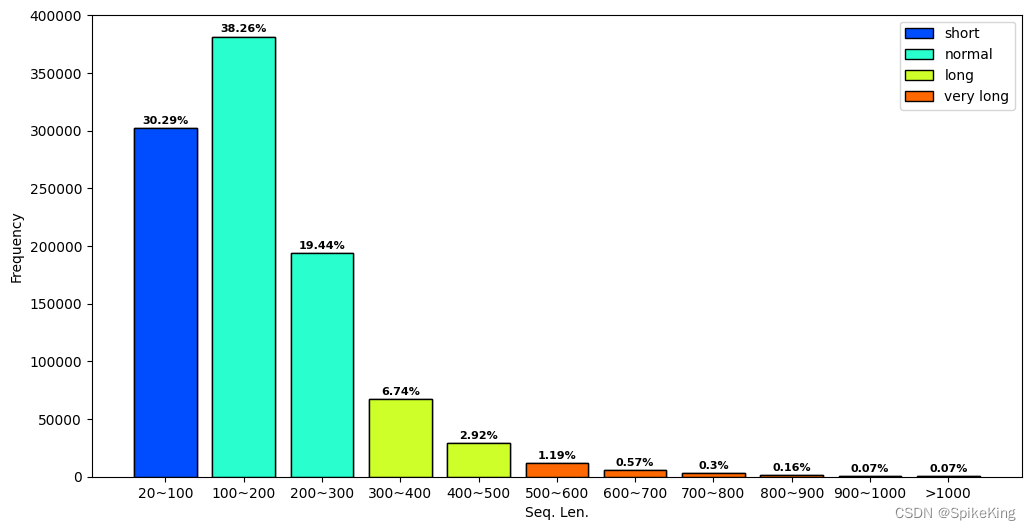
4. Highquality Clust 30 聚类合并
执行步骤:
- 遍历全部文件夹,统计样本数:36987928,即约3700万个PDB,目录文件4.1G,遍历一次约1.5h。
- 使用
highquality_clust30.fasta聚类,输入36997632个PDB,比真实样本多9704个,输出22668950,预估9294269个高质量样本,即929万。 - 数据处理时间,提取pLDDT的值,以及序列,大约1周。
安装MMseqs2:
conda install -c conda-forge -c bioconda mmseqs2
再次进行聚类,设置--min-seq-id 0.3,聚类命令如下:
# 第1次聚类, 由 36997632 下降为 22668950
mmseqs easy-linclust highquality_clust30.fasta clusterRes tmp --min-seq-id 0.3 --kmer-per-seq 100 --cluster-mode 2 --cov-mode 1 -c 0.8 --db-load-mode 2
执行日志:
createdb highquality_clust30.fasta tmp/15932757348140161575/input --dbtype 0 --shuffle 1 --createdb-mode 1 --write-lookup 0 --id-offset 0 --compressed 0 -v 3
Shuffle database cannot be combined with --createdb-mode 0
We recompute with --shuffle 0
Converting sequences
[36997614] 34s 585ms
Size of the sequence database: 36997632
Size of the alignment database: 36997632
Number of clusters: 31407973
Size of the sequence database: 31407973
Size of the alignment database: 31407973
Number of clusters: 22668950
...
输出3个文件:
clusterRes1_all_seqs.fasta
clusterRes1_cluster.tsv
clusterRes1_rep_seq.fasta # 聚类输出文件
参考
- How to Extract (Unzip) Tar Gz File
- StackOverflow - How to check python anaconda version installed on Windows 10 PC?
- 编程随笔 - 服务器配置 Conda 和 Jupiter Lab 的环境
- linux大文件压缩及解压需要注意问题
- CSDN - No module named ‘torch_geometric‘解决办法
- 序列聚类(mmseqs2)
- How could I “recreate” UniRef50/UniRef90 with MMSEQS2?
- GitHub - easy-linclust # 420