原文标题:Regression Transformer enables concurrent sequence regression and generation for molecular language modelling
论文地址:Regression Transformer enables concurrent sequence regression and generation for molecular language modelling | Nature Machine Intelligence
代码地址: https://github.com/IBM/regression-transformer
1、思想
学习输入和目标变量的联合分布可以模糊预测与条件生成模型之间的界限(统一预测模型与条件生成模型), permutation language modelling (PLM)模型同时擅长回归与条件序列生成。
目前只有ChemFormer同时解决了这两个问题,但是需要调整特定任务头,因此没有真正提出一个多任务模型。现有的transformer模型要么调整特定的任务头,要么将两者之间通信限制为奖励/损失(图a),从而无法完成回归与结构生成。性质驱动的生成模型应当擅长性质识别(图bcdef)。
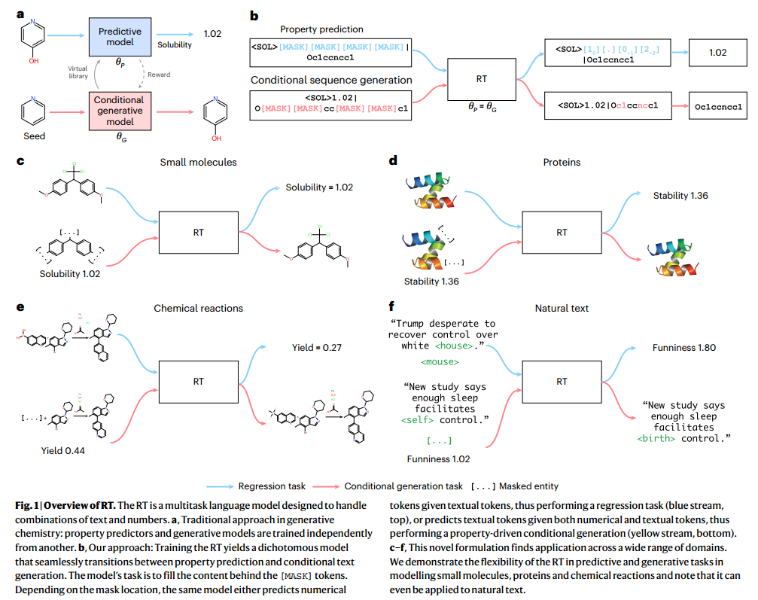
2、模型方法
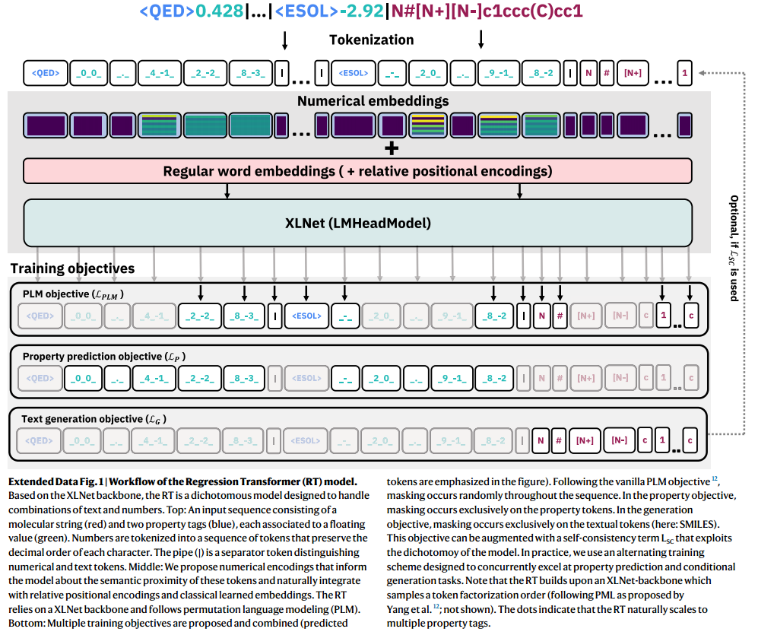
1)模型架构选择(XLNet backbone):
语言模型包括:自回归 (RNN、GPT)或者MLM (BERT)。自回归适合长序列,但是不能应用于文本填充和从MLM中获益。MLM不适合生成长序列。因此选择综合模型XLNet作为backbone,其主要是自回归模型,但是设置了特殊的训练目标,因此可以获得双向关注。如BERT双向模型中的独立性假设随着<mask>被逐步填充,其破坏性也随着增加(因为前面的都预测准确后,后面一旦错误,前面预测的都是白费的),特别是基于骨架的“装饰”,由于大量被mask,导致单个原子的生成会严重影响分子功能。所选择的模型应当:受益于具有完整序列关注的自回归生成
2)Tokenization:
数字:以前对于指定性质的生成都是整数(8-bit),此处用任意浮点精度表示实数。由于数字的稀疏性(一旦指定一个数,就变得稀疏了),将单个数字作为单个token是次优的,因此将数字的回归转化为序列的分类任务(实际上是将数字按正则表达式拆分设计为字符串):
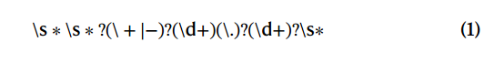
每个能够匹配上的数字字符被转化为一个token Tv,p ,其中v为数字,p为位置,如12.3 → [1_2,2_0.,3_-1]
字母:对于SMILES,转化为SMLFIES,并使用固有的tokenizer
蛋白质序列:氨基酸token
3)Numerical Encodings (NE)
由于数字的固有结构,以纯粹数据驱动的方式学习数字token embedding可能是无效的。(应该是作者定义的数字固有结构,去直接学习每个token是可能无效的)。此外由于RT模型是交叉熵损失训练的(典型回归模型),因此没有传达数字token之间的相似性概念。因此提出NEs(类似于位置编码),给定一个token Tv,p,其N_dim为j,
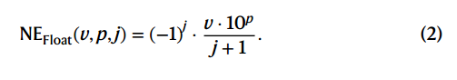
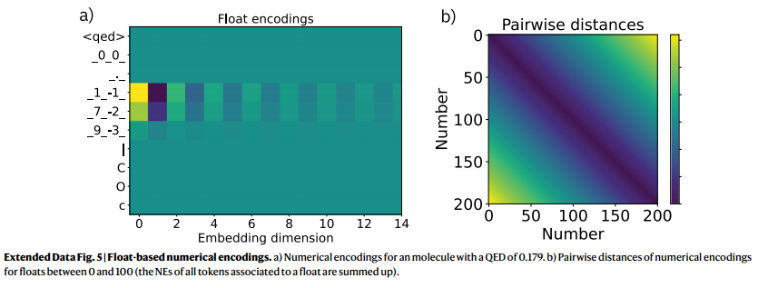
因此NE的振幅随着数值的变化而变化(NE在百位上与千位上存在一个数量级上的振幅,没有数字的位子NE设为0)。同时表示正负数之间的接近性(在NE上只相差一维)。但是NE在奇数-偶数维度上的正负交替在高纬度上逐步消失(图5a 表明正负在高纬度上并没有啥影响)。关键是,成对距离是对称的,并随着浮点值单调衰减(图5b,???这说明了啥)
4)Training objectives
输入包括k property tokens + l textual tokens ,为:
![]()
全长 T = k + l。
PLM objective.
思想:通过在运行时对序列x的因式分解顺序z采样来自回归地填充masked token。根据分解顺序分解似然pθ(x),期望得到一个双向自回归模型:
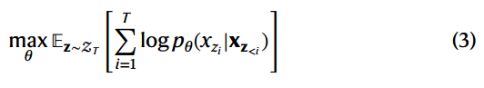
在实际中,只部分预测(预测最后c个token)。因此损失可以改为(c为一个超参数):
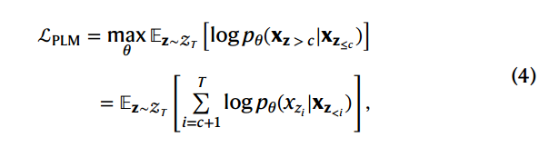
通常每个batch中,masked ratio = 1/c . 注意这里没有对property tokens 、 textual tokens做区分,形式上通用,但是实际中需要排列(堆叠后就仅仅对一个),因此需要特殊处理:交替训练(设置两个目标,分别用于性质预测、文本预测),使得模型可以预测数字标记或者文本标记。
Property prediction objective.
屏蔽全部性质token,就是将前c = l个token mask,因此损失为:
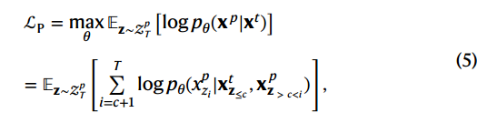
注意:x向量排列顺序变为(文本token,性质token)
Conditional text generation objective.
在给定性质token和部分文本token的情况下促进了masked文本token的生成(即重建全文序列),即前k个token不mask,设置c≥k,保证只mask文本:
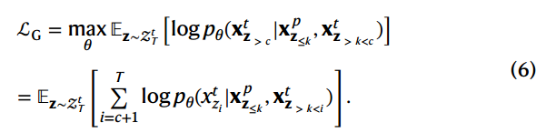
注意:x向量排列顺序变为(性质token,文本token)
Self-consistency (SC) objective.
如果生成的序列符合primed性质,则上述(6)不会奖励(即损失不会再次下降),单个符号(即原子、氨基酸或(子)词)的变化可以极大地改变序列(句子)的性质(含义),添加一个SC term(第二项)来补充:
![]()
直观地说,它是样本的性质与生成样本的预测性质之差。
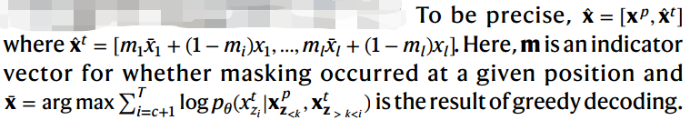
在这样的公式中,RT在其自身优化过程中充当了一个约束层(类似于自监督的额外层)。奖励与训练样本不同的分子的生成,只要符合属性。
3、Model training and evaluation procedure
Implementation
HuggingFace库XLNet作为backbone,添加了一个额外的编码层用于预训练heads,数字Ndim = 16 (即j = 16),在Transformer encoder 32个隐藏层, feed-forward的维度为256和1024,16个heads(20% dropout)。大约2700万个可训练的参数(确切的数字取决于词汇量的大小)。单个(NVIDIA Tesla A100或V100)
Chemical language modelling.
药物相似度(QED)。从ChEMBL72中的约160万个生物活性分子开始,通过使用RDKit计算所有分子的QED分数(q∈[0,1])并舍入到小数点后三位。约140万训练,1000个验证,10000个测试。使用公式4训练模型,直到验证集收敛,然后训练公式5、7每隔50步交替。SMILES/SELFIES的词库量为509/724。在评估中,greedy decoding用于性质预测,beam search decoding用于分子生成。设置c=2.5(大约40%的token的被mask,maximum span: seven tokens)
MoleculeNet benchmark.
ESOL, FreeSolv and Lipophilicity。使用15%的验证数据执行三次随机分割。使用QED初始化热启动,并只训练了50,000step(batchsize=4)。由于QED预训练使用了[0,1]中的数值,将整个MoleculeNet数据集的值归一化到相同的范围(仅使用训练数据),并将它们四舍五入到小数点后三位。对于所有目标,除非另有约束,设置屏蔽c = 5,并将连续屏蔽令牌的跨度限制为最多5 tokens。
Property optimization benchmark.
目标是调整种子分子,使其属性最大化,同时固定的相似性约束,其中215,381个训练和799个测试。pLogP是辛醇-水分配系数(logP),由 synthetic accessibility score和大于6个原子的循环数决定。因此,pLogP可以像QED一样从分子中确定地计算出来。QED初始化进行热启动,训练50,000step,用相同的种子分子prompting 80次,但改变分数和掩码的最大跨度,形成一个分子池
Protein sequence language modelling.
蛋白质相互作用指数(Boman)。专注于波曼指数(衡量肽的潜在蛋白质相互作用的指标)。从UniProt中收集了包含15-45个氨基酸的全部2,648,205条肽,计算了它们的Boman指数,并分别使用10,000和1,000进行测试和验证。
最初在[−3.1,6.1]范围内的Boman指数数值被归一化为0,1(仅使用训练数据),并四舍五入到小数点后三位。一组用于PLM目标(方程(4)),两组用于交替目标。再次在优化(方程(5)和方程(7))之间每隔50step交替进行,并训练一组有SC损失的模型和一组没有SC损失的模型(方程(7)中的α分别= 1和α = 0)。
TAPE benchmark.
Fluorescence and Stability数据集。目标是分别预测与训练蛋白相差一到四个突变的蛋白质的荧光和内在折叠稳定性。这两个数据集都有固定的分割。荧光(稳定性)数据集有21,446(53,416)个训练样本,5,362(2,512)个验证样本和27,217(12,851)个测试样本。
使用Boman初始化(PLM目标)热启动三个模型,并训练直到验证性能饱和(~100,000步),数值再次缩放到[0,1]。
对于条件生成任务的评估,模型被赋予了更大的灵活性:60%的令牌被屏蔽(即式(3)中的c = 1.7),最大跨度为7个氨基酸残基。用于预训练的Boman数据集包含15-45个残差(mean±s.d)。(36±7),荧光蛋白显著增加(246±0.2个残基,P < 0.001)。相反,稳定性数据集中的蛋白质大小与预训练数据相似(45±3个残基)。
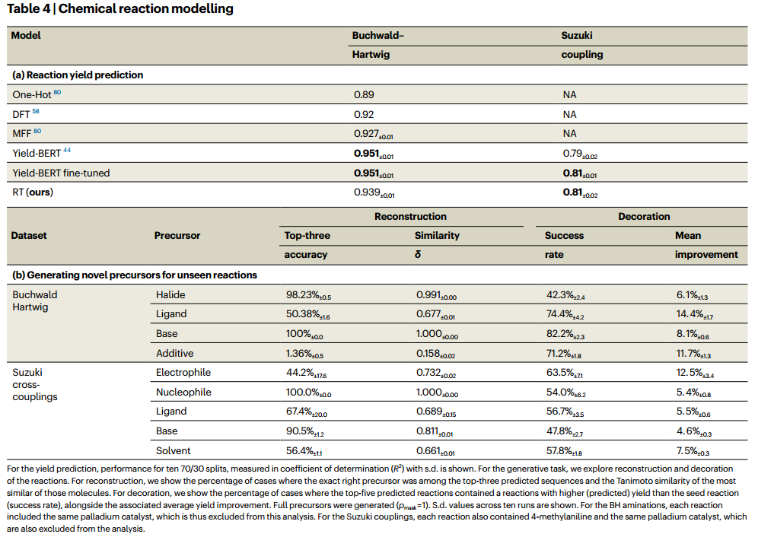
Chemical reaction modelling.
USPTO数据集。由于这两个反应产率数据集只覆盖了化学空间的狭窄区域(一个模板应用于许多前体组合),对从USPTO中提取的更广泛的反应化学模型进行了预热。总共有5000个反应进行验证,并对模型进行训练,直到方程(5)和方程(7),α = 1上的验证性能饱和。c设为2.5。reaction SELFIES 的词汇包含861个token。
Reaction yield datasets.
10个模型分别在重复的随机分裂上进行微调。10个模型分别在重复的随机分裂上进行微调。训练目标再次在属性预测(方程(5))和条件生成(方程(7),α = 1)之间每隔50步交替进行,最多50,000步
4、Results
1、Chemical language modelling
Initial validations—learning drug likeness.
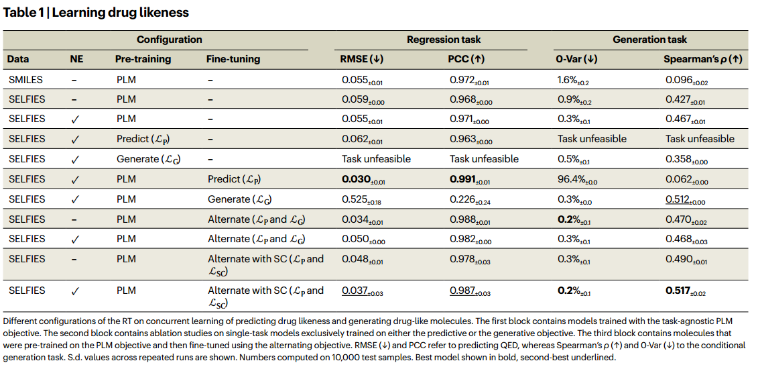
对于生成任务,验证分子进行10次查询,其间隔为[0,1]和40%的掩码文本标记。“引物”的意思是用期望的性质值代替序列的真实性质。高秩相关ρ(引物与唯一生成分子的QED之间)值表明该模型成功地完成了损坏序列以生成具有所需QED的完整分子。值得注意的是,所有模型的新颖性得分>99%。RT可以产生新的化学物质,坚持一个连续的性质感兴趣。
NEs(类似于位置编码),在所有任务中都略微提高了性能
回归任务甚至实现了均方根误差(RMSE) <0.06
对于单目标可以取得较好表现,但是当任务难度逐渐增加(即seed分子的QED与primed QED的距离)时,生成分子的QED与引物属性的距离线性增加),因为构成了一个局外分布(模型只用seed == primed预训练)。
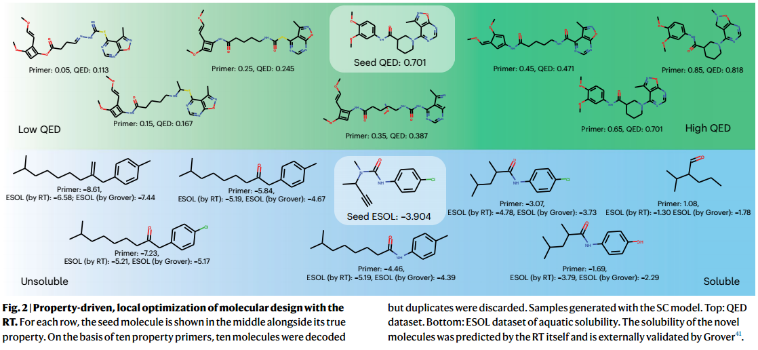
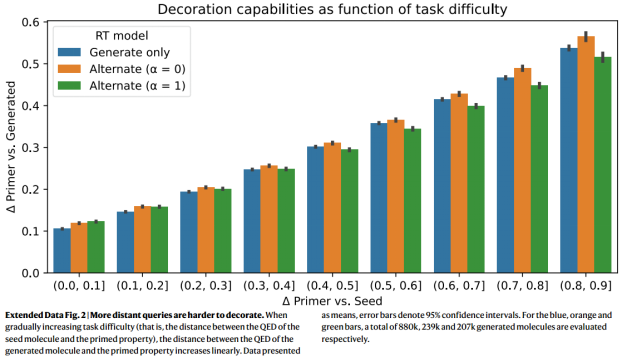
SELFIES模型远远超过了SMILES模型,因为SMILES在语法上是无效的(发现60%的有效性)。然而,这个数字很难与非种子生成模型进行比较,因为(1) RT必须修复损坏的SMILES,不能简单地依赖其自身的内部状态;(2)同时提供的属性引物捕获了从低到高的QED分数的整个范围,从而激励模型冒险地装饰序列以坚持约束属性-这一任务通常是不可能的,并且很容易导致broken SMILES; (3) RT训练不依赖于教师强迫。虽然SELFIES有1.6%损坏,但是相对而言很低了
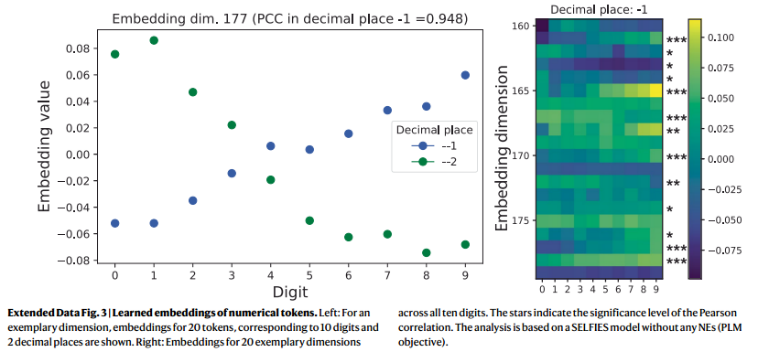
Learning embeddings of numbers.
NEs对于QED的提升很小。如扩展数据图3所示,在没有 static NEs的情况下,模型从数据中学习数字的自然顺序。大量的嵌入维数(小数点- 1和- 2分别为47%和36%)直接和显著地编码了数字的顺序(即,10个嵌入值与严格单调向量之间的P < 0.05和∣PCC∣>0.62)。例如,在扩展数据图3(左)中,数字值与其嵌入值单调相关。
2、Regression benchmark (MoleculeNet)
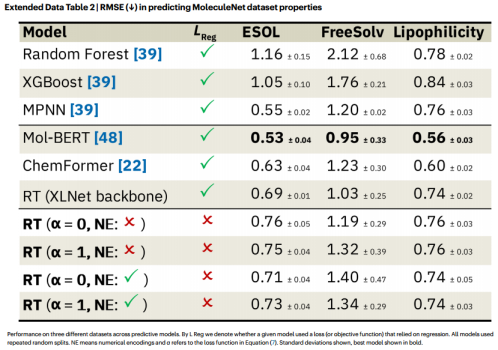
MoleculeNet的最强基线模型XGBoost在所有任务上都优于我们的所有模型。我们所有的模型都优于BERT和BART。值得注意的是,这些模型在微调回归头之前利用了大规模的自监督预训练,而我们使用的是分类损失。由于这些结果可能无法直接与带有XLNet主干的RT进行比较,因此我们还对带有传统回归头的XLNet模型进行了微调。值得注意的是,尽管没有回归损失,RT是相同的(亲脂性)或只是轻度较差(即在sd范围内;ESOL, FreeSolv)到XLNet。
3、Conditional molecular generation benchmark
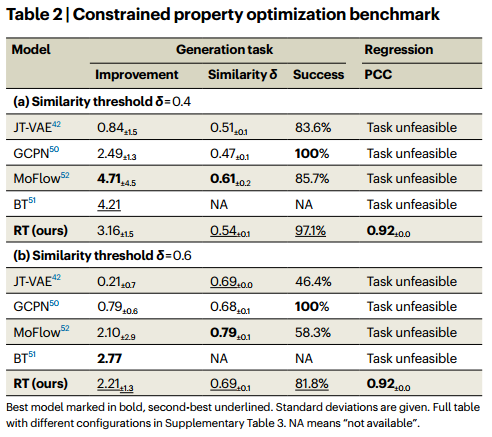
given in Tanimoto similarity 阈值δ,生成高plogP, RT竞争的最先进的模型。这与RT训练形成鲜明对比,RT训练只有在重建分子具有与种子分子相似的(预测的)plogP时才会获得奖励(我们没有为训练构建定向plogP查询;它们仅在推理时使用)。因此,RT在价态上是不可知的,同样可以用来使分子适应较低的plogP。RT能够在目标定向分子生成中与专门的条件生成模型竞争。同时,RT还预测了plogP值,Pearson相关系数(PCC)为0.92,这是常规条件生成模型无法解决的任务。
4、Protein sequence language modelling
Pre-training on potential protein interaction (Boman index).
RT成功地生成了具有所需的波曼指数的肽,给定部分损坏的氨基酸序列(斯皮尔曼ρ为0.84;表3 b)。此外,更高比例的掩码令牌在蛋白质生成任务中可以获得更好的结果。
TAPE datasets (protein fluorescence and protein stability).
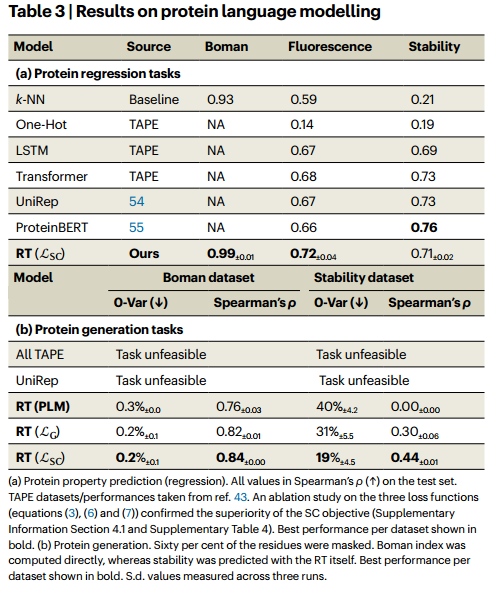
RT的竞争性预测性能表明,自监督预训练的好处可以扩展到数字标记的数据集。这产生了一个条件生成模型,用于蛋白质序列空间的属性驱动的局部探索。关于这一点的证据可以在表3b中找到:尽管所有的TAPE模型以及UniRep方法都无法解决这一生成任务,但RT能够修改测试蛋白,使其(预测的)稳定性与引物特性强烈相关(ρ = 0.44)。
5、Modelling chemical reactions