AMBER分子动力学模拟之分子模拟-- HIV蛋白酶-抑制剂复合物(2)
体系平衡
体系的优化分为两步,第一步固定蛋白和小分子,对添加的水盒优化。第二步则是对整个体系的优化。
对水盒子优化
vim min1.in
&cntrl
imin=1,
maxcyc=10000,
ncyc=5000,
ntb=1,
ntr=1,
ntpr=500,
cut=10,
ioutfm=1,
ntxo=1,
restraint_wt=500.0,
restraintmask=':1-199',
/
对复合物优化
vim min2.in
&cntrl
imin = 1,
maxcyc = 50000,
ncyc = 25000,
ntb = 1,
ntr = 0,
ntpr = 500,
cut = 10,
ioutfm = 1,
ntxo = 1,
/
升温
由于动力学模拟的是真实的生物体环境,因此必须使研究对象升温升压到临界值,体系达到平衡,才能做实际的动力学模拟
vim md0.in
&cntrl
imin=0,
irest =0,
ntx=1,
ntb=1,
cut =10,
ntr=1,
ntc =2,
ntf=2,
tempi=0.0,
temp0=300.0,
ntt =3,
gamma_ln=1.0,
nstlim=150000,
dt=0.002,
ntpr=500,
ntwx=1000,
ntwr=500,
restraint_wt=10.0,
restraintmask=':1-199',
/
动力学模拟
一般来说第一步的MD模拟时间要长一些,确保体系构象已经稳定,之后进行第二步的MD用于能量、相互作用分析等。
vim md1.in
&cntrl
imin=0,
irest=1,
ntx=5,
ntb=2,
pres0=1.0,
ntp=1,
taup=2.0,
cut=10,
ntr=0,
ntc=2,
ntf=2,
tempi=300.0,
temp0=300.0,
ntt=3,
gamma_ln=1.0,
nstlim=20000000,
dt=0.002,
ntpr=500,
ntwx=500,
ntwr=500,
/
vim md2.in
&cntrl
imin=0,
irest=1,
ntx=5,
ntb=2,
pres0=1.0,
ntp=1,
taup=2.0,
cut=10,
ntr=0,
ntc=2,
ntf=2,
tempi=300.0,
temp0=300.0,
ntt=3,
gamma_ln=1.0,
nstlim=5000000,
dt=0.002,
ntpr=500,
ntwx=500,
ntwr=500,
ntwprt=3198,
/
mdrun 执行文件
在mdrun.exe文件中,调用amber的pmemd.cuda模块进行动力学模拟
vi mdrun.exe写入
### CPU
sander -O -i min1.in -p pep.top -c pep.crd -o min1.out -r min1.rst -ref pep.crd < /dev/null
sander -O -i min2.in -p pep.top -c min1.rst -o min2.out -r min2.rst < /dev/null
sander -O -i md0.in -p pep.top -c min2.rst -o md0.out -x md0.crd -r md0.rst -ref min2.rst < /dev/null
sander -O -i md1.in -p pep.top -c md0.rst -o md1.out -x md1.crd -r md1.rst < /dev/null
sander -O -i md2.in -p pep.top -c md1.rst -o md1.out -x md2.crd -r md2.rst < /dev/null
### cpu_mpi
mpirun -np 2 sander -O -i min1.in -p pep.top -c pep.crd -o min1.out -r min1.rst -ref pep.crd < /dev/null
mpirun -np 2 sander -O -i min2.in -p pep.top -c min1.rst -o min2.out -r min2.rst < /dev/null
mpirun -np 8 sander -O -i md0.in -p pep.top -c min2.rst -o md0.out -x md0.crd -r md0.rst -ref min2.rst < /dev/null
mpirun -np 8 sander -O -i md1.in -p pep.top -c md0.rst -o md1.out -x md1.crd -r md1.rst < /dev/null
mpirun -np 8 sander -O -i md2.in -p pep.top -c md1.rst -o md1.out -x md2.crd -r md2.rst < /dev/null
## GPU
pmemd.cuda -O -i min1.in -p pep.top -c pep.crd -o min1.out -r min1.rst -ref pep.crd < /dev/null
pmemd.cuda -O -i min2.in -p pep.top -c min1.rst -o min2.out -r min2.rst < /dev/null
pmemd.cuda -O -i md0.in -p pep.top -c min2.rst -o md0.out -x md0.crd -r md0.rst -ref min2.rst < /dev/null
pmemd.cuda -O -i md1.in -p pep.top -c md0.rst -o md1.out -x md1.crd -r md1.rst < /dev/null
pmemd.cuda -O -i md2.in -p pep.top -c md1.rst -o md2.out -x md2.crd -r md2.rst < /dev/null
目前准备已经准备好进行md的参数文件(带有水盒子信息的拓扑和坐标文件)、md的输入文件,min1.in min2.in md0.in md1.in md2.in,以及MD执行文件mdrun.exe
将mdrun.exe的权限设置为可执行文件
chmod +xmdrun.exe
nohup./mdrun.exe&
输出文件
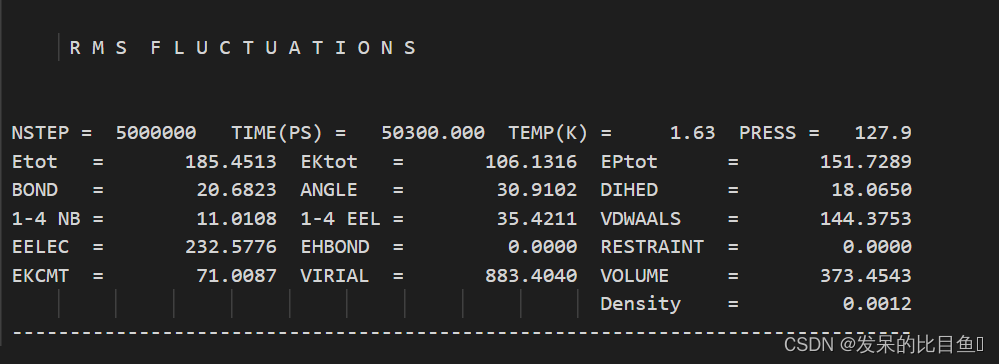
在md的输出文件中,所有的*out文件均含有每一步相对应的Amber版本,工作日期,工作路径、输入文件的详细参数,包含未曾修改的默认参数、引用文献、我们重点关注和分析的是每一步输出的能量项等等。
vi min1.out
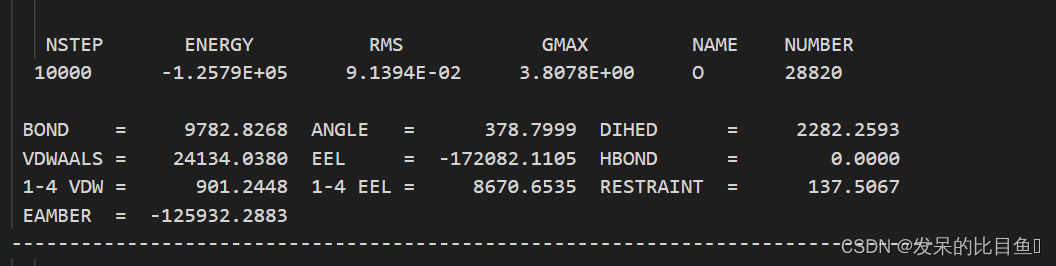
vi min2.out
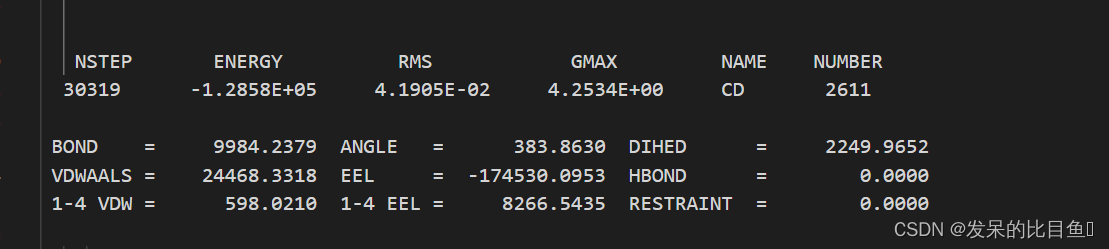
vi md0.out
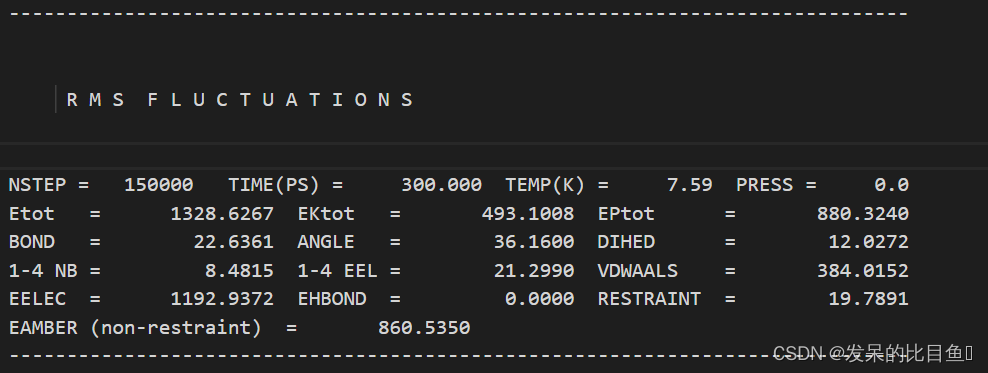
vi md1.out
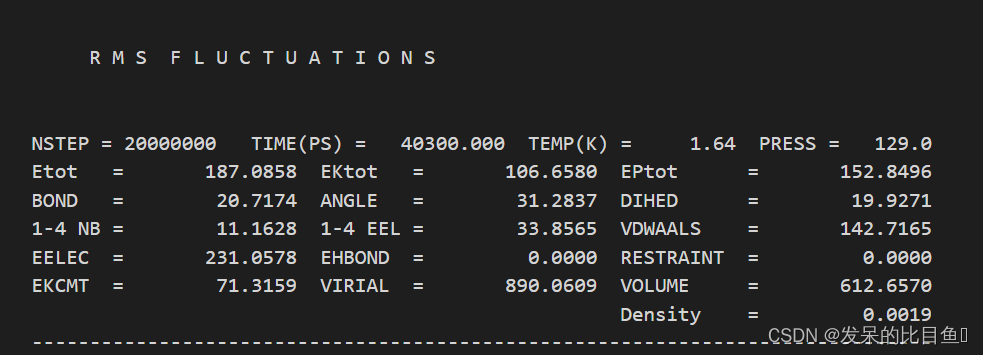
vi md2.out