数量性状基因座作图原理与步骤
经典的数量遗传分析方法 ->只能分析控制数量性状表现的众多基因的综合遗传效应,无法准确鉴别基因的数目、单个基因在染色体上的位置和遗传效应
(1)数量性状基因座(QTL)
Quantitative trait loci: QTL 数量性状位点(基因座)
- 所谓QTL是指通过连锁分析来确定位于遗传标记附近的染色体区域中的某些可以确定其位置、数目与效应的控制数量性状的基因.
(2)QTL作图
- 一个数量性状往往受到多个QTL影响,这些QTL分布于整个基因组的不同位置
- 利用特定的遗传标记 鉴定控制数量性状的QTL 在基因组中的位置、数目及其遗传效应的过程,称之为QTL作图(QTL mapping),亦称QTL定位
- QTL作图的目的:确定某数量形状受到多少个QTL控制,了解其所在染色体并估计其对数量性状作用的效应
- 以QTL定位为核心的分子数量遗传学研究发展迅速.
QTL作图的基本原理
利用特定遗传分离群体中的遗传标记及相应的数量性状观测值,分析遗传标记和形状之间的连锁关系.
如果分析结果证明某个遗传标记与性状连锁,则可认定在该标记附近存在一个或几个QTL。分析一个性状与已知连锁图的一系列标记之间的连锁关系,即可确定存在多少个QTL及这些QTL在标记图谱上的位置
需要注意的是:QTL作图中的连锁分析与质量性状不同,不能直接计算遗传标记和QTL之间的重组率,而是采用统计学方法计算它们之间连锁的可能性(LOD值),依据这种可能性是否达到某个阈值来判断遗传标记和QTL是否连锁,并进而确定其位置和效应.
下面会解释遗传标记是什么
以单标记和单基因为例子
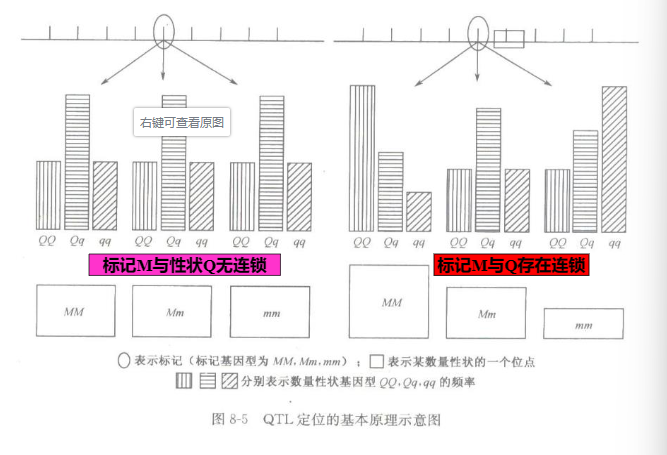
- 若标记M与性状Q无连锁(左图),则不同的M标记基因型(MM、Mm、mm)所对应的Q基因型(QQ、Qq、qg)比例分布相同(均遵循孟德尔规律),因此3种M标记基因型所对应的Q性状平均值会相等.
- 若标记M与Q存在连锁(右图),不同的M标记基因型所对应的Q基因型比率分布会受M标记连锁影响发生改变,因此3种M标记基因型所对应的Q性状在平均数上有差异,这是数量性状定位的最基本原理.
QTL定位的基本步骤
(1)选择遗传标记
理想的作图标记应该具有4个方面的特征:
- 数量丰富:标记覆盖整个基因组
- 多态性好:个体或亲代与子代之间有不同的基因型
- 中性:同一基因位点的各种基因型都有相同的适应性,以避免不同基因型之间的生存能力差异引起的试验误差.
- 共显性:以保证直接区分同一基因位点的各种基因型.形态标记数量有限,通常不表现中性和共显性.
蛋白质标记可以满足中性和共显性,但它们又有数量不足或多态性不好的缺点,而DNA分子标记容易具备上述4个特征,已成为目前应用最广泛的作图标记。
常用的DNA分子标记

(2)人工构建作图群体
适于QTL定位的群体应该是待测数量性状存在广泛变异,多个标记为点处于分离状态的群体,这种的群体一般是由亲缘关系较远的亲本间杂交,再经自交回交等方法进行人工构建的.如用高株×矮株或早熟期×晚熟期等,常用的群体有:
- F2群体
- 回交(BC)群体
- 双单倍体(doubled haploids,DH,即加倍的单倍体群体)群体
- 重组近交系 (recombinant inbred lines,RL,由F,连续多代自交产生) 群体
其中DH群体和RL群体的分离单位是品系,品系间存在遗传差异而品系内个体间基因型相同,自交不分离,可以永久使用。
重组近交系群体:是杂种后代经过多代自交得到的一系列高世代家系。由于自交的遗传效应使纯合个体增多,杂合个体减少,最后获得家系内基因型纯合稳定一致、家系间基因型不同的RILS永久性群体是研究分子标记的理想材料。应用纯合的后代群体进行QTL定位,不需要很大的鉴定群体。
(3)获得某数量性状的表型与遗传标记基因型的相关数据
从作图群体中抽样(每株)提取DNA做分子标记检测,记录每个被测个体的标记基因型。若标记的遗传图谱未知,还需要先依据各标记基因型分离资料制作标记的连锁图.
- 如西红柿质量(大小)差异很大的两个纯系亲本的杂交.

(4)测量数量性状
在检测作图群体的每个个体的标记基因型值的同时,测定其数量性状值。将每个个体的数量性状表现型值和分子标记基因型值按顺序列表,就形成了后续分析的基本数据

(5)统计分析
- 用统计方法分析数量性状与标记基因型值之间是否存在关联,判断QTL与标记之间是否存在连锁(用Mapmaker / Exp3.0软件进行连锁遗传分析),确定QTI在标记遗传图谱上的数目、位置,估计 QTL的效应
简而言之,QTL定位一般包括以下步骤:

QTL区间定位
利用两个相邻的分子标记检测和定位处于其间的QTL,即所谓QTL区间定位法。该方法的运用需要借助最大似然法及其相关的LOD值来进行数据处理和判定
似然 (likelihood) 是一个统计学概念。用于处理某种观测结果出现的可能性,而最大似然法则是用以满足其估计值在观察资料的结果中出现的概率最大,此方法又称最大似然估计。在QTL的区间定位中,需要计算出某QTL在两个相邻的分子标记区间中各位置的LOD值,而LOD值是似然比L/L的常用对数来表示的,即

其中:L=基因座之间存在连锁的概率,亦即表示两个标记物之间的重组率r相连锁的概率。
L0=不存在连锁的概率,亦即统计学中的所谓“零假设”进行QTL区间定位时,用LOD值作为判断某染色体某区域存在QTL与否的阈值,当LOD值在2或3时,则表明在检测区间内某QTL效应显著,则可确定存在一个QTL与遗传标记连锁。
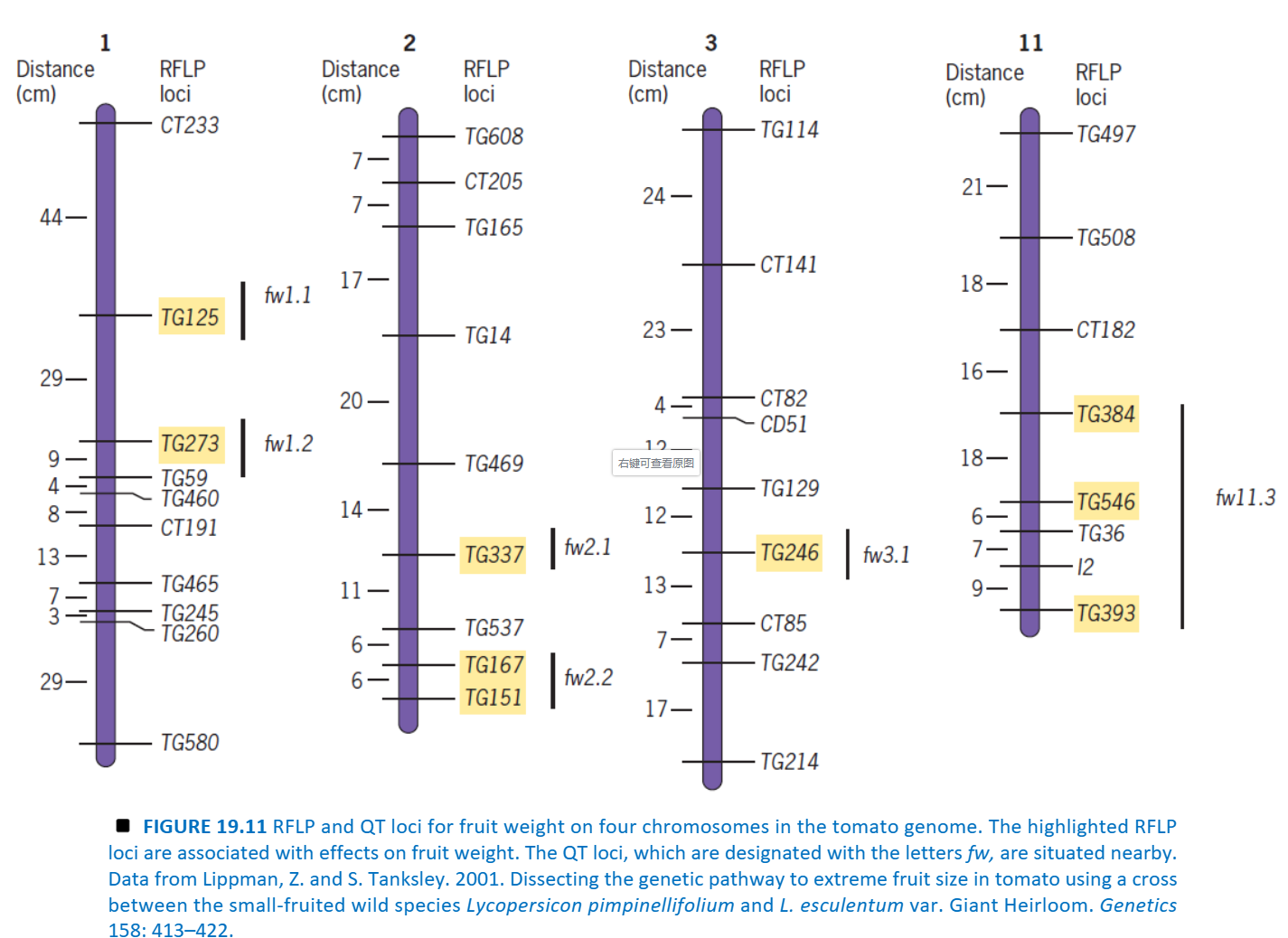
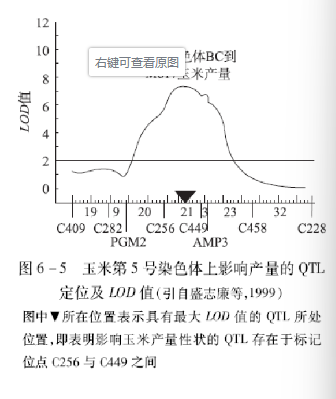
现以玉米的两个近交系:B73xMo17的F1×Mo17回交群体(BC)的试验为例说明影响其产量性状的QTL区间定位结果。
采用同工酶 (PGM2,AMP3) 和RFLP标记 (C409,C282,C256,C449.C458,C228)共计8个标记位点;各标记间的距离为厘摩 (cM) ;LOD值为2的水平线是该试验的阈值
QTL分析的应用前景
QTL分析的应用主要有三个方面:
- 由QTL定位得到的遗传图谱可以进一步转换成物理图谱,对QTL进行克隆和序列分析,在DNA分子水平上研究决定数量性状基因的结构和功能,进而应用基因工程的手段来操纵QTL。
- 用于标记辅助选择(Marker-assisted Selection,MAS ) 。在动植物育种上,利用标记与QTL的连锁,在实验室内对数量性状变异提早进行识别与选择,可以提高选择效率和精度。与传统方法相比,标记辅助选择对回交育种引入隐性有利基因、剔除非轮回亲本不利连锁基因更为快速有效,可以较少世代数完成目的基因的转育
- 利用标记与OTL连锁分析可以提供与杂种优势有关的信息,鉴定与杂种优势有关的标记位点,确定亲本在QTL上的差异,可以有效地预测。
希望能帮到你~





![[Data structure]单链表 | 一文介绍线性数据结构之一的单链表(Java实现)](https://img-blog.csdnimg.cn/f4eedcae0aa84f9084a0ac893f9494e1.png)