1写在前面
不知道大家那里天气热了没有,苦逼的我虽然“享受”着医院的恒温,但也并没有什么卵用,毕竟我只是个不可以生锈的“小螺丝”。🥲
上期介绍了Scillus包的基本功能,如何进行数据的预处理及其可视化。🤨
本期我们继续吧,介绍其主要的一些高级功能吧,包括降维,统计,热图和富集分析等等。🤩
2用到的包
rm(list = ls())
library(tidyverse)
library(Scillus)
library(Seurat)
library(magrittr)
library(purrr)
3示例数据
我们用一下上次经过标准化处理后的scRNA_int,具体的可以回顾上一篇教程:👇
📍🧐 Scillus | 来吧!它可以大大简化你的Seurat分析流程哦!~(一)(数据预处理)
load("./scRNA_scillus.Rdata")
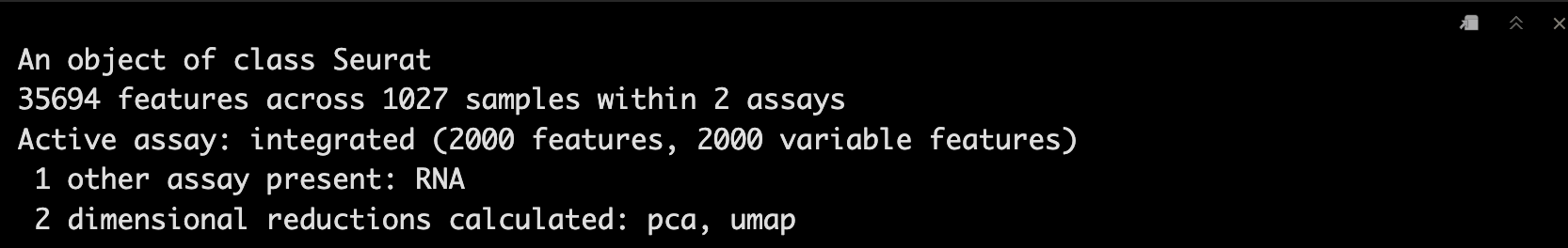
scRNA_int
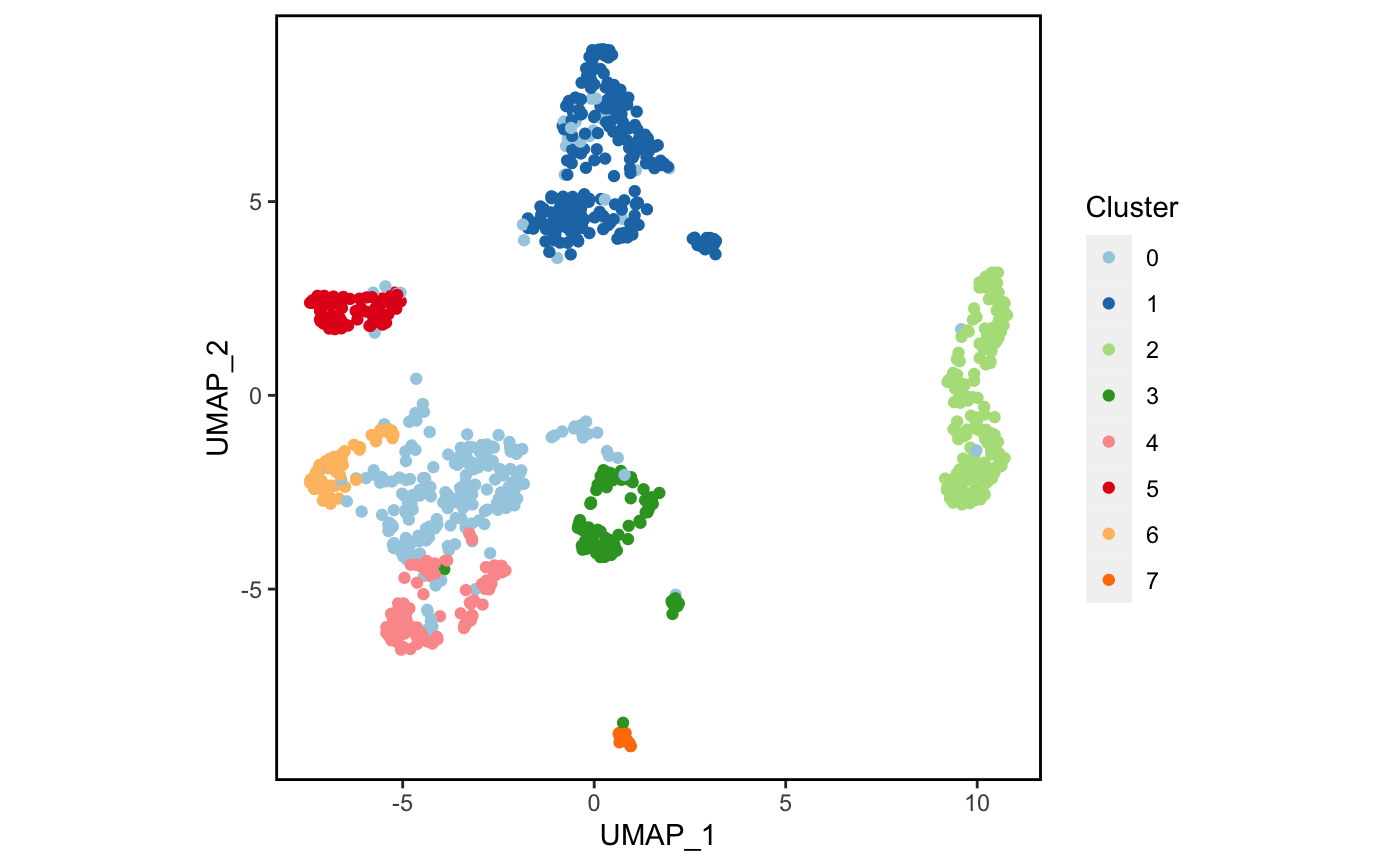
4降维及其可视化
4.1 初步绘图
先来简单画一下吧。🥳
plot_scdata(scRNA_int, pal_setup = pal)
4.2 换个主题配色
换个配色试试。🥰
plot_scdata(scRNA_int, pal_setup = "Dark2")

4.3 分组可视化
我们试试分组降维吧。🤒
plot_scdata(scRNA_int, color_by = "group", pal_setup = pal)
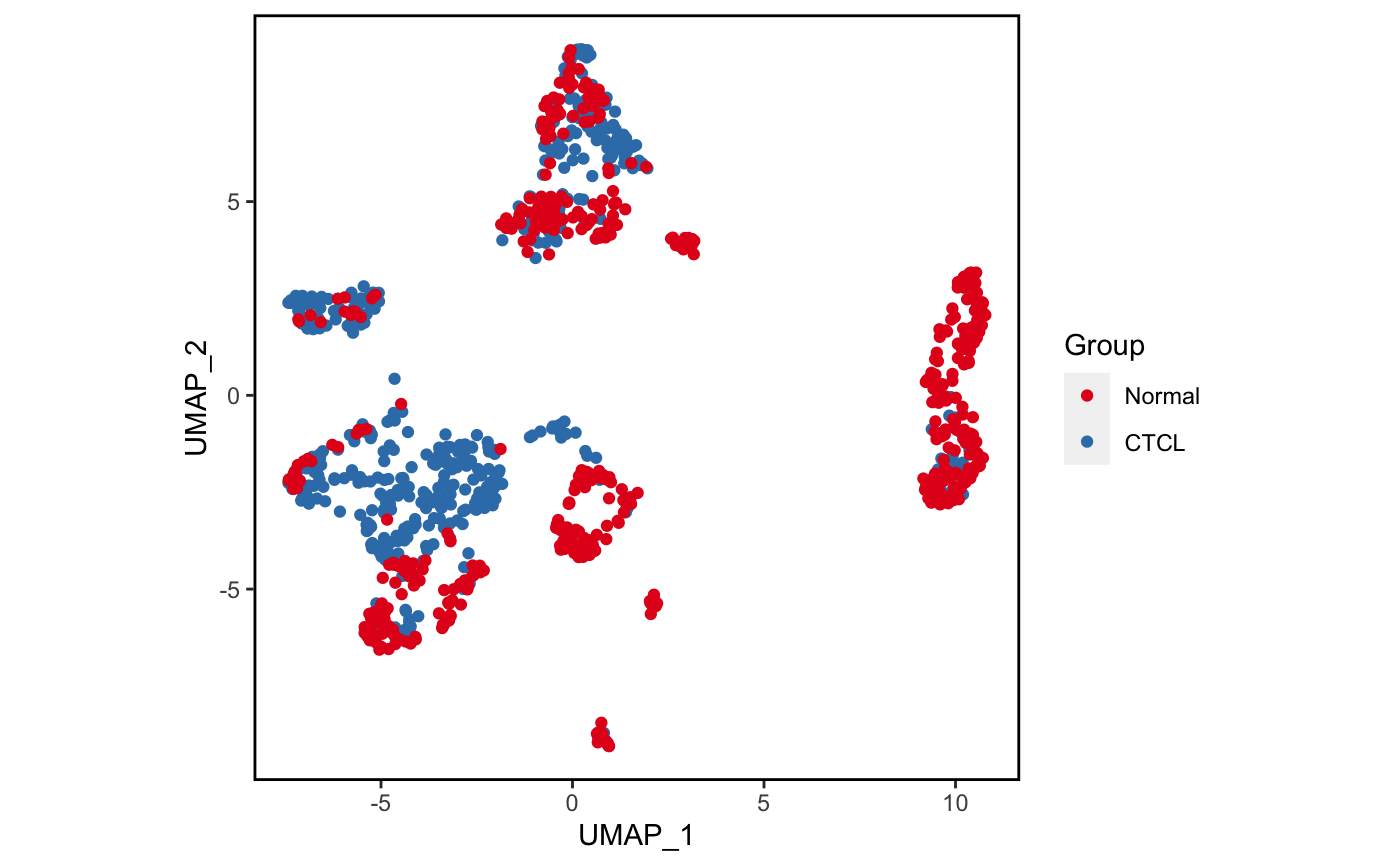
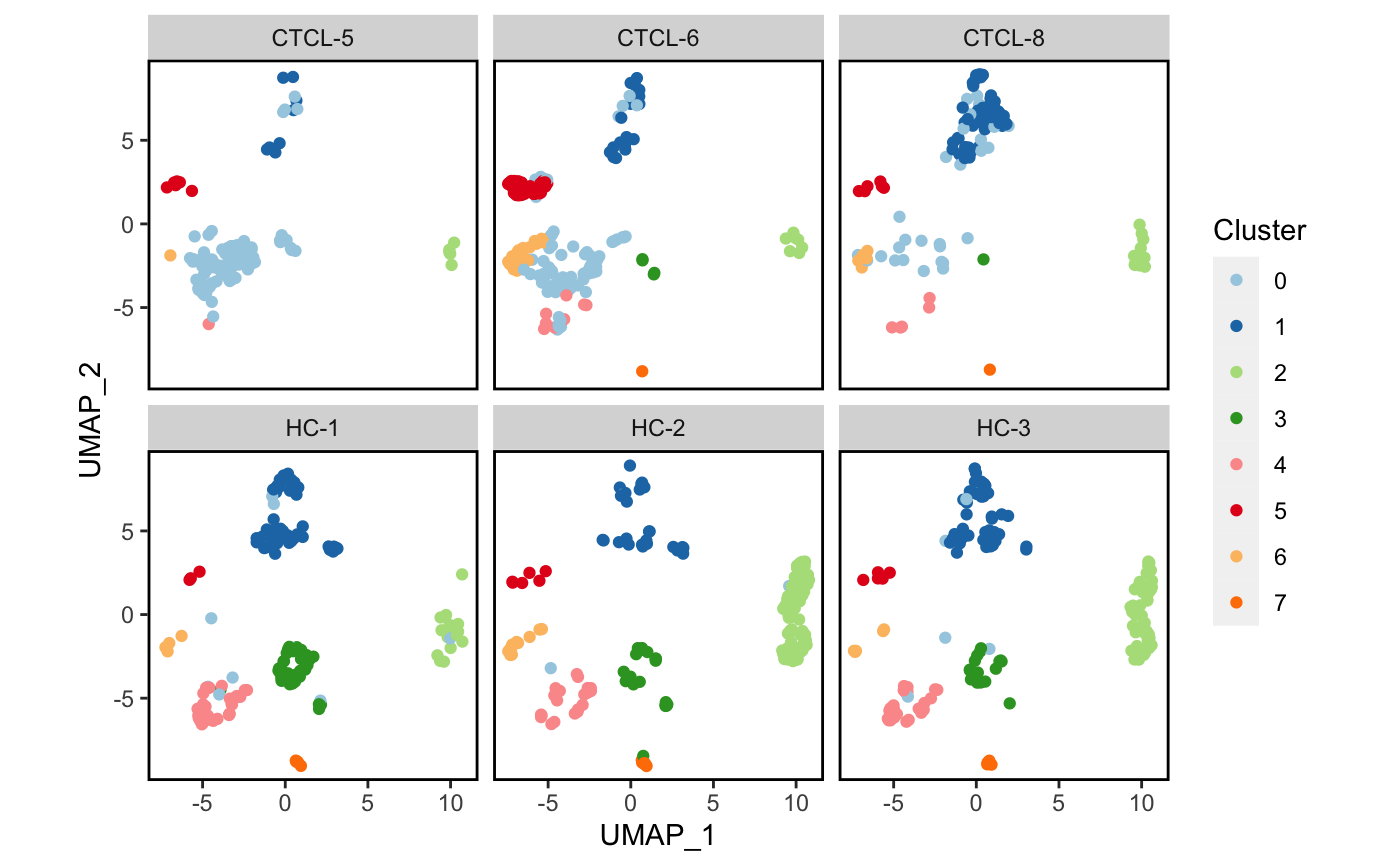
4.4 分面可视化
我们试一下按sample分面降维吧。🤒
plot_scdata(scRNA_int, split_by = "sample", pal_setup = pal)
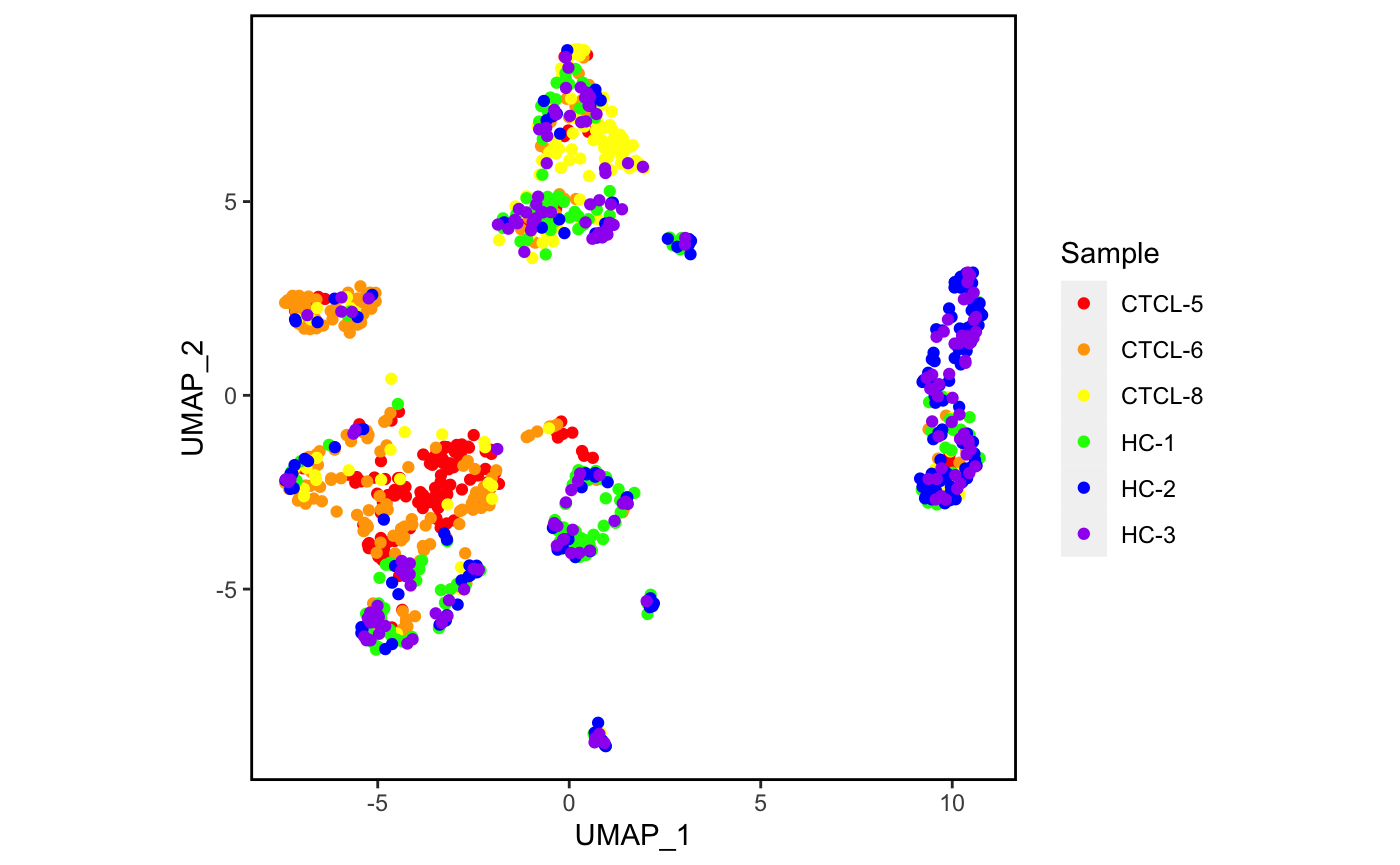
4.5 手动配色
我们再试着手动配色一下吧。🧐
plot_scdata(scRNA_int, color_by = "sample",
pal_setup = c("red","orange","yellow","green","blue","purple"))
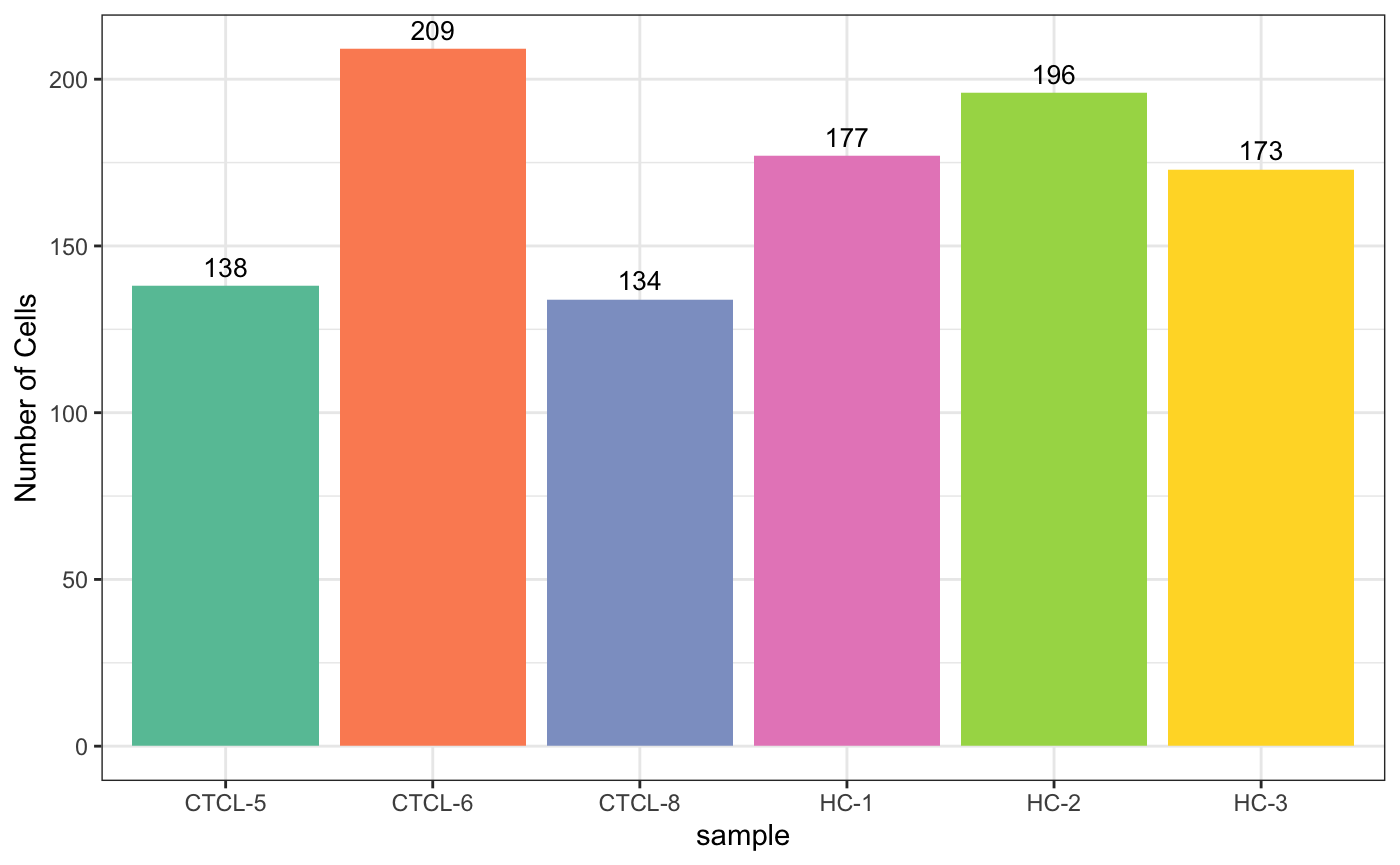
5统计及其可视化
5.1 按sample统计
plot_stat(scRNA_int,
plot_type = "group_count"
## 三种,"group_count", "prop_fill", and "prop_multi"
)
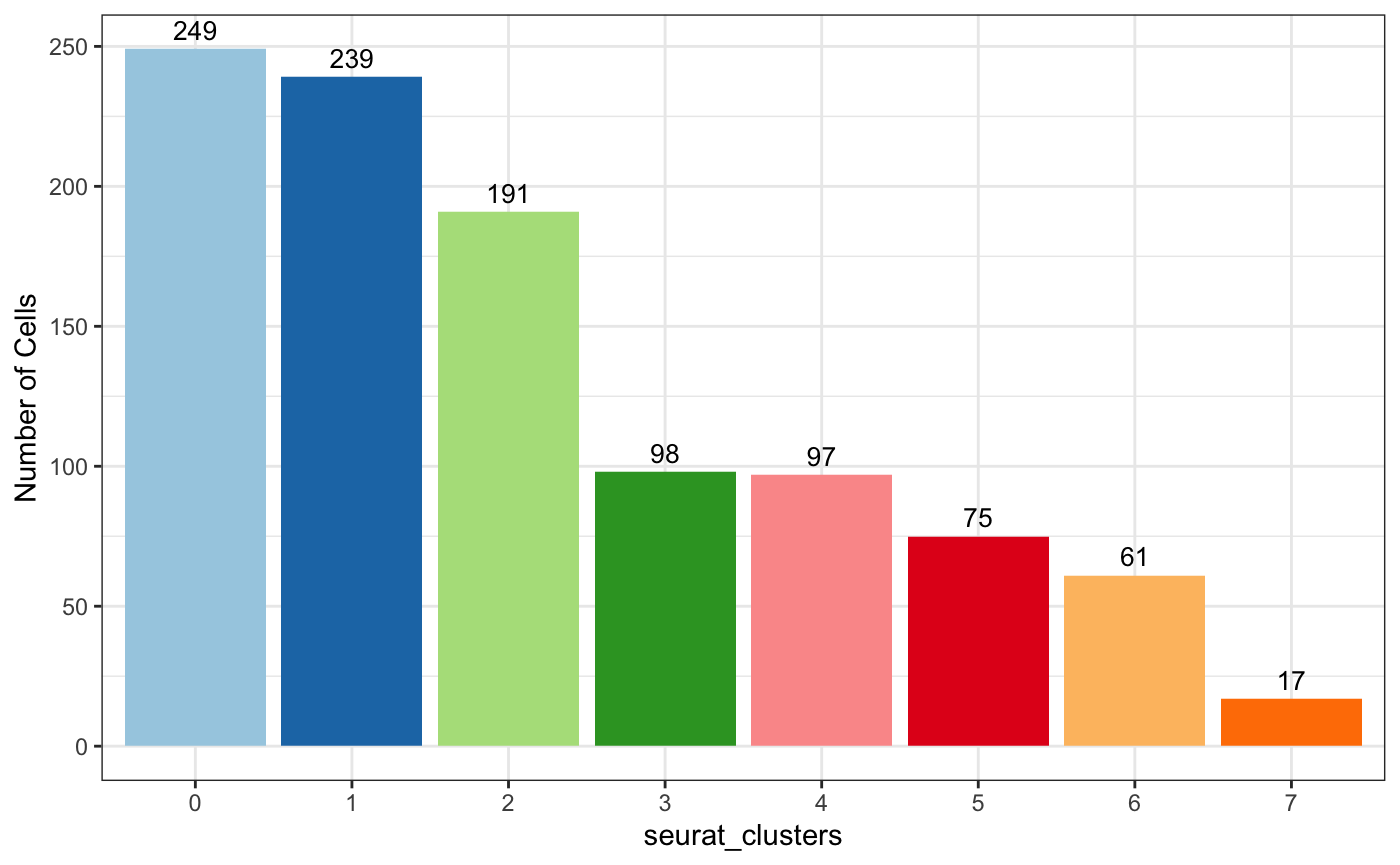
5.2 按cluster统计
plot_stat(scRNA_int, "group_count", group_by = "seurat_clusters", pal_setup = pal)
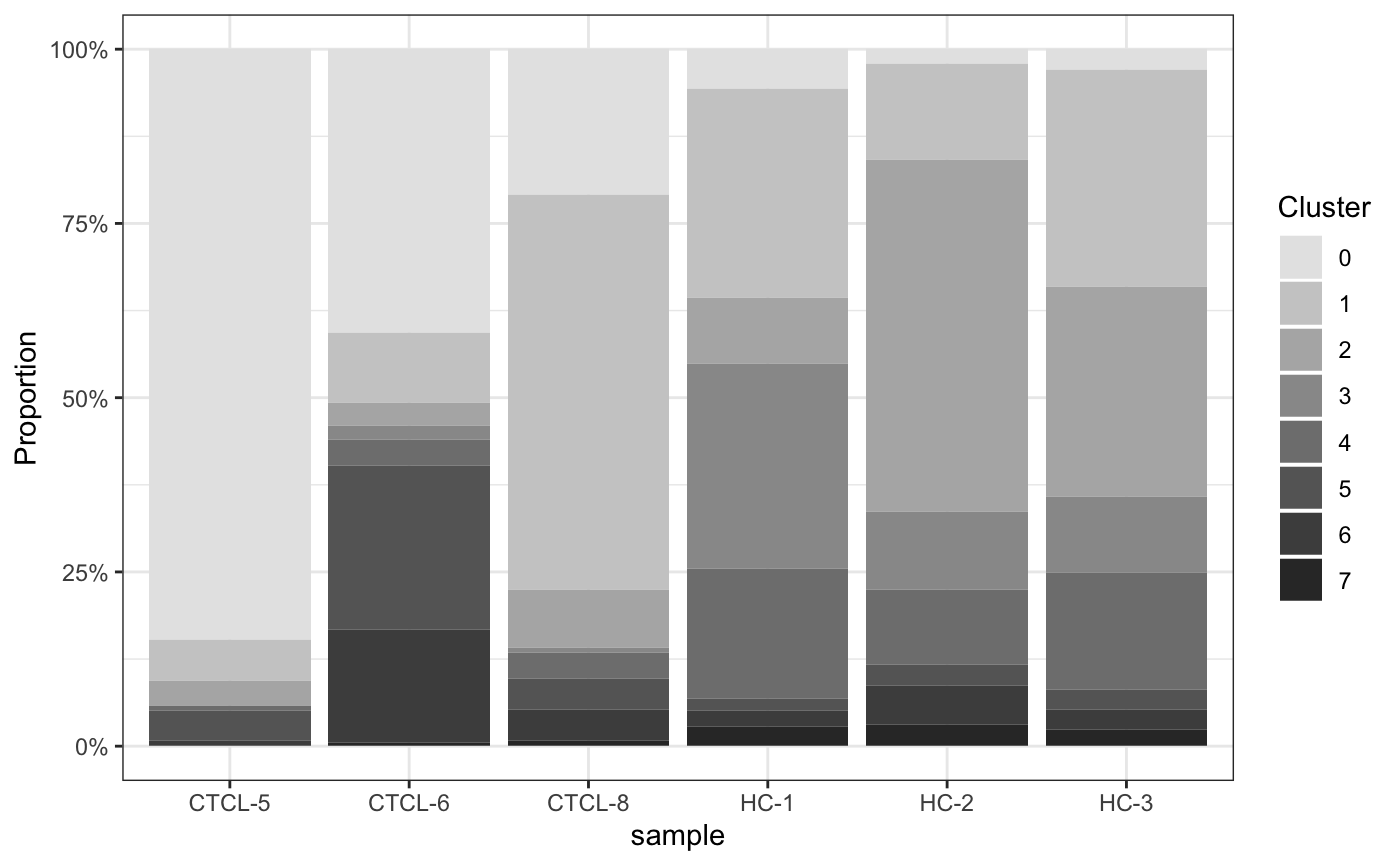
5.3 堆叠柱形图
plot_stat(scRNA_int,
plot_type = "prop_fill",
pal_setup = c("grey90","grey80","grey70","grey60","grey50","grey40","grey30","grey20"))
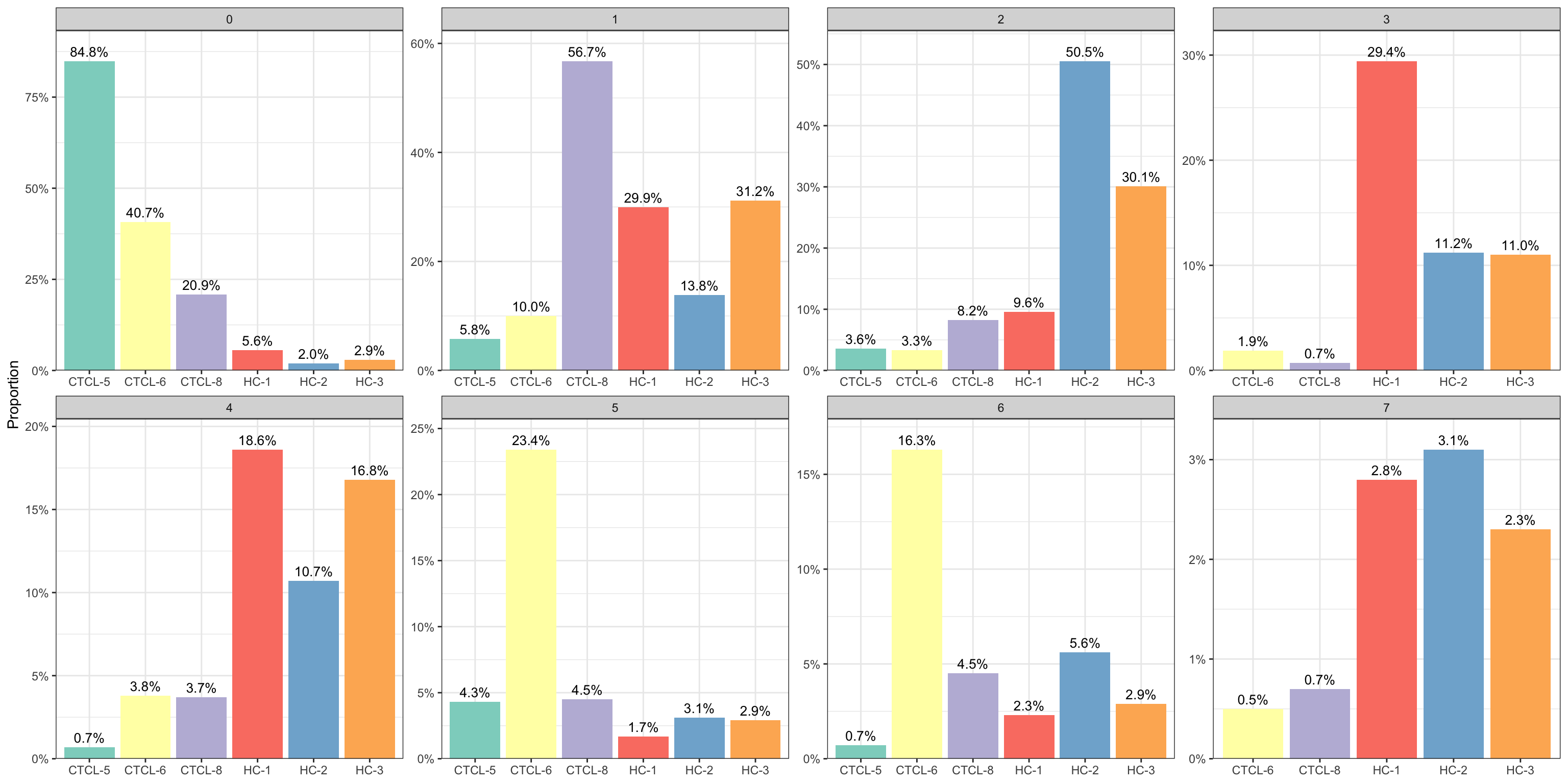
5.4 按cluster和sample统计
plot_stat(scRNA_int, plot_type = "prop_multi", pal_setup = "Set3")
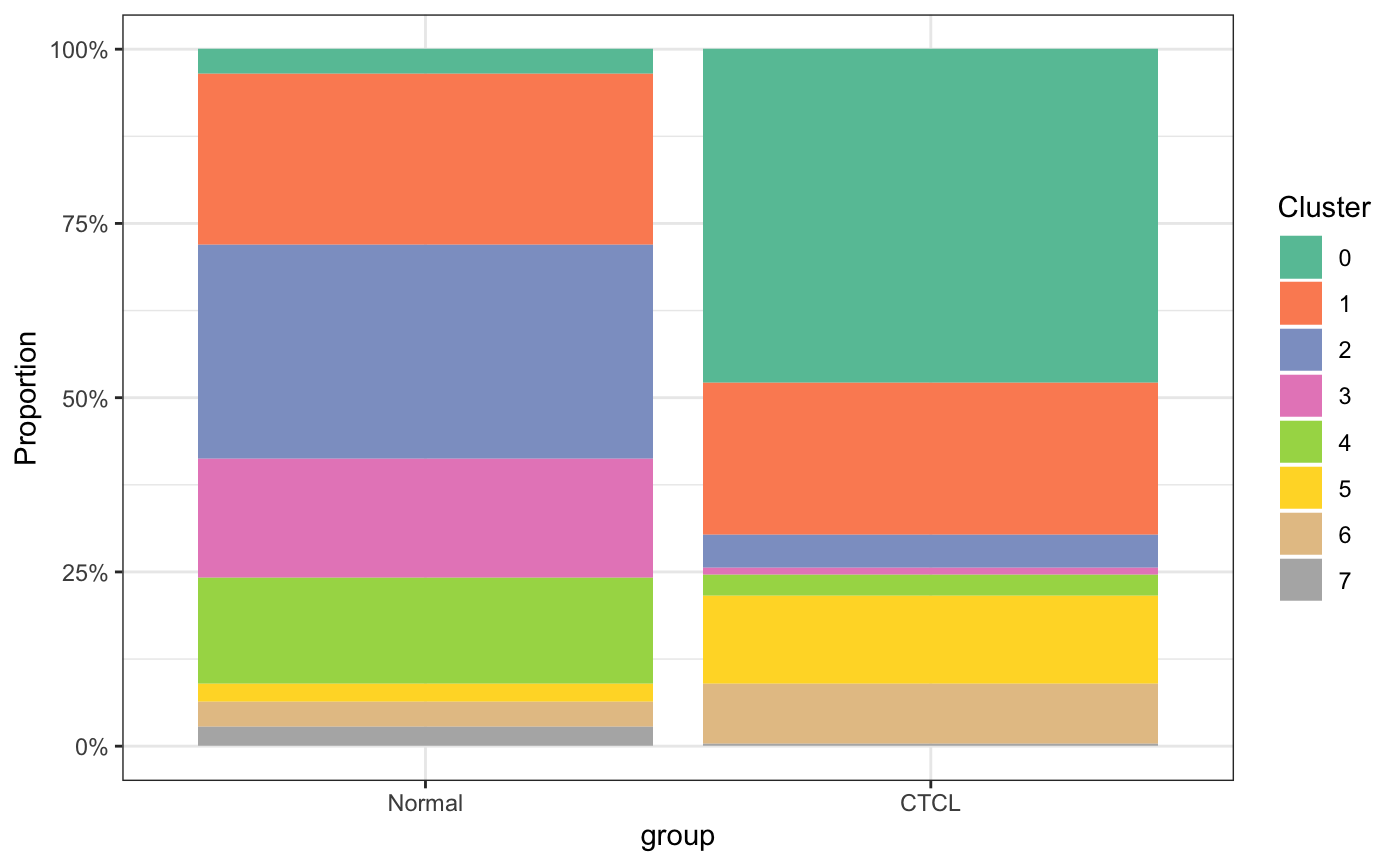
5.5 按cluster和group统计
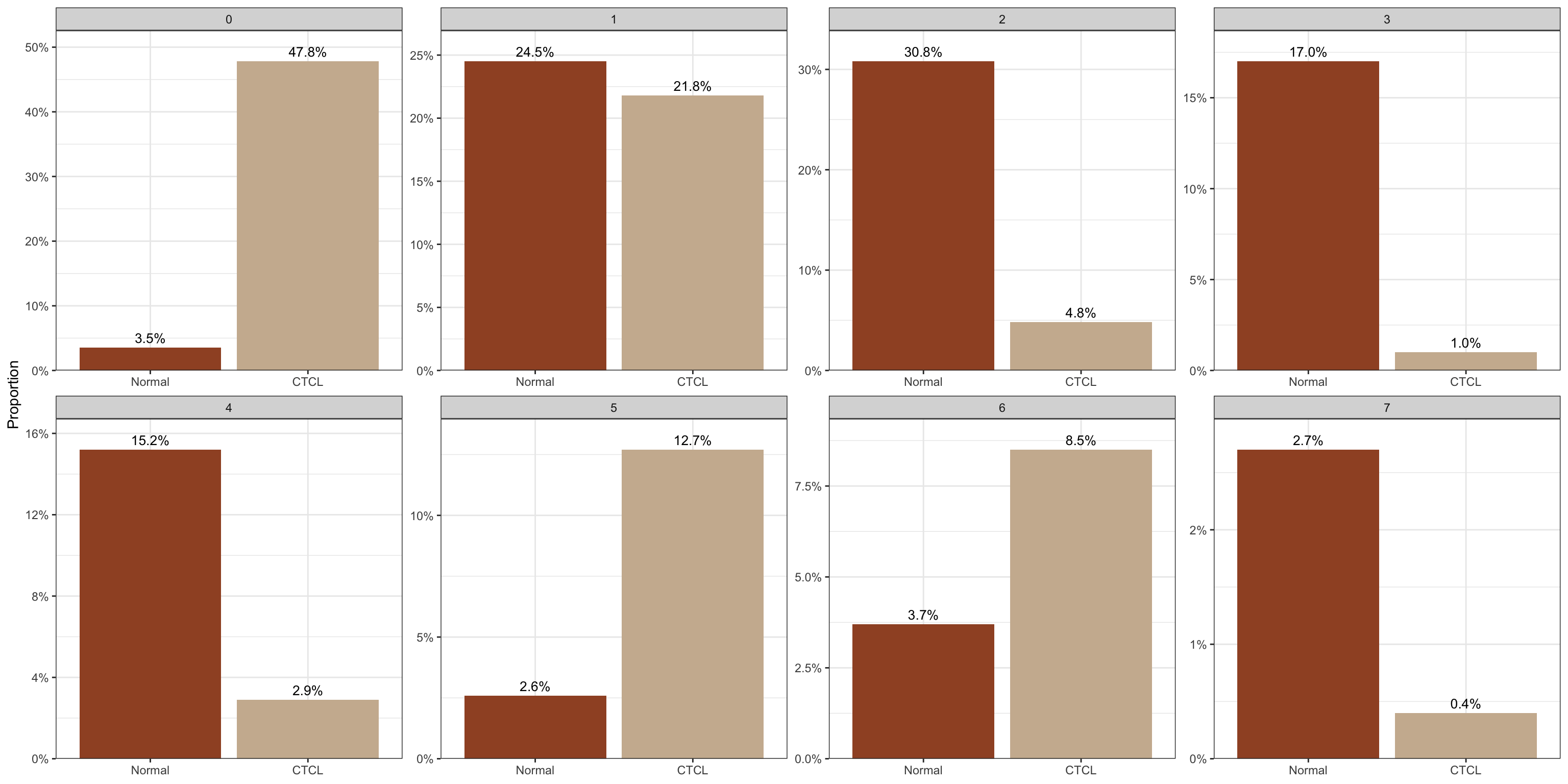
plot_stat(scRNA_int, plot_type = "prop_fill", group_by = "group")
5.6 换个配色
plot_stat(scRNA_int, plot_type = "prop_multi",
group_by = "group", pal_setup = c("sienna","bisque3"))
6热图及其可视化
6.1 寻找marker
我们首先要用Seurat包的FindAllMarkers确定一下marker,分析方法也比较多,包括wilcox, roc, t, poisson, DESeq2等。😷
markers <- FindAllMarkers(scRNA_int, logfc.threshold = 0.25, min.pct = 0.1, only.pos = F)
6.2 热图可视化
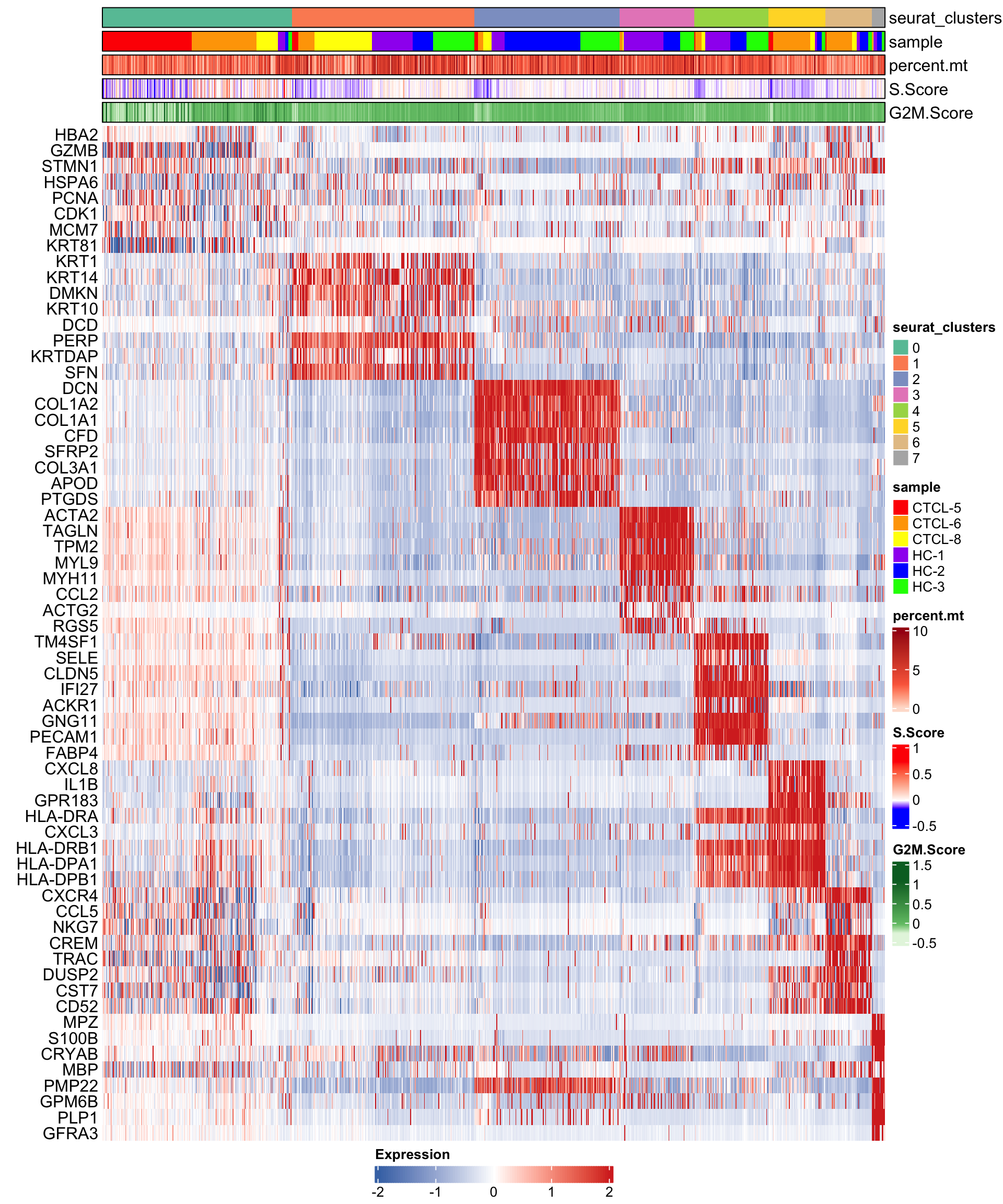
在热图中,每一行代表一个基因,每一列代表一种细胞,默认绘制每种细胞的前8个基因。🤓
细胞可以通过sort_var进行排序,默认设置为c("seurat_clusters"),即细胞按cluster进行排序。🙃
当然你也可以在sort_var中指定多个变量,然后按多个变量进行排序。🥳
anno_var用来指定注释数据或者metadata中的相关数据。🥰
plot_heatmap(dataset = scRNA_int,
markers = markers,
sort_var = c("seurat_clusters","sample"),
anno_var = c("seurat_clusters","sample","percent.mt","S.Score","G2M.Score"),
anno_colors = list("Set2",
# RColorBrewer palette
c("red","orange","yellow","purple","blue","green"),
# color vector
"Reds",
c("blue","white","red"),
# Three-color gradient
"Greens"))
6.3 调整热图
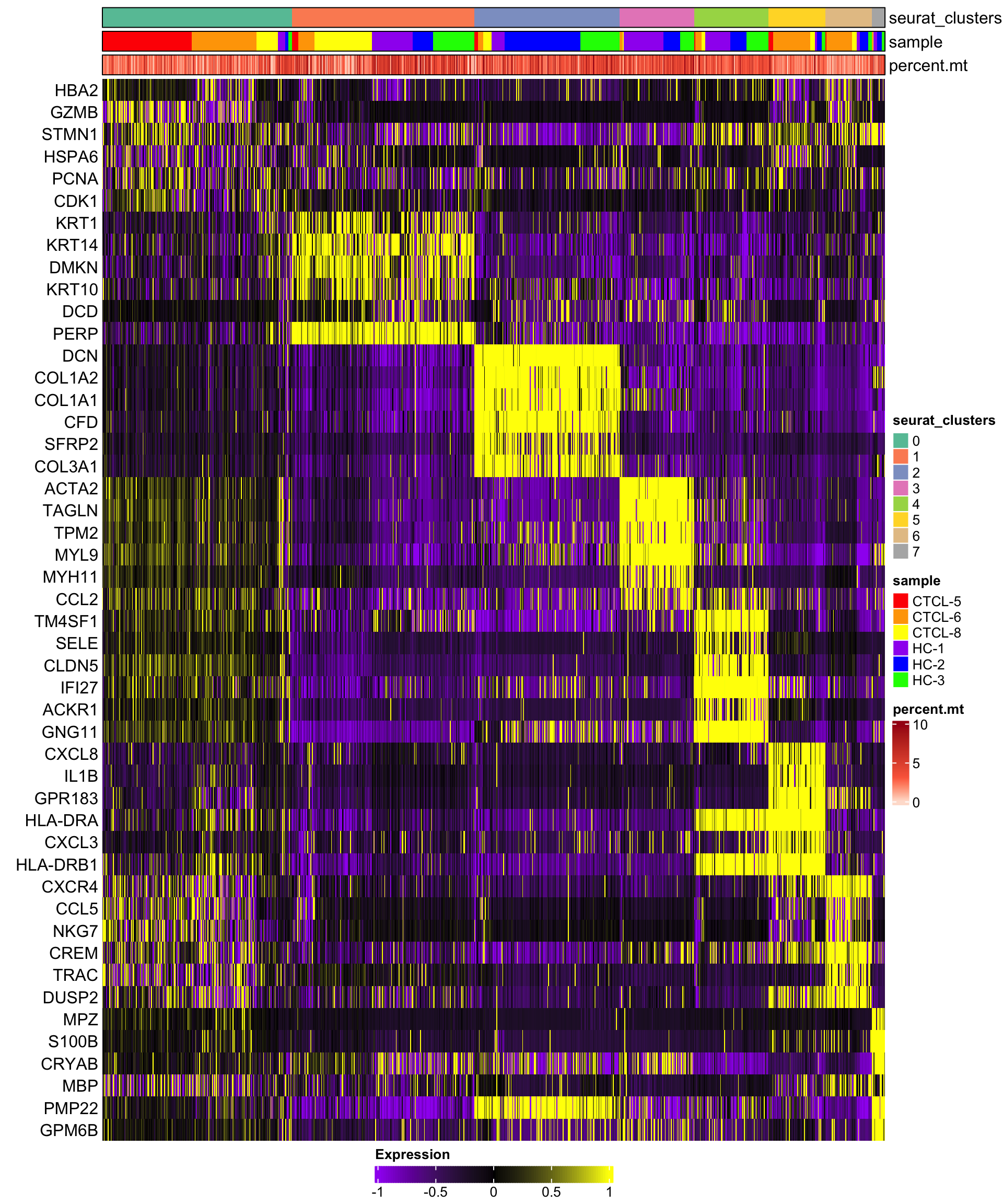
我们调一下limit和渐变的配色, 显示前6个基因。😂
plot_heatmap(dataset = scRNA_int,
n = 6,
markers = markers,
sort_var = c("seurat_clusters","sample"),
anno_var = c("seurat_clusters","sample","percent.mt"),
anno_colors = list("Set2",
c("red","orange","yellow","purple","blue","green"),
"Reds"),
hm_limit = c(-1,0,1),
hm_colors = c("purple","black","yellow"))
7富集分析
7.1 制定cluster的GO分析
我们指定cluster 1的GO分析试试。🐵
plot_cluster_go(markers, cluster_name = "1", org = "human", ont = "CC")
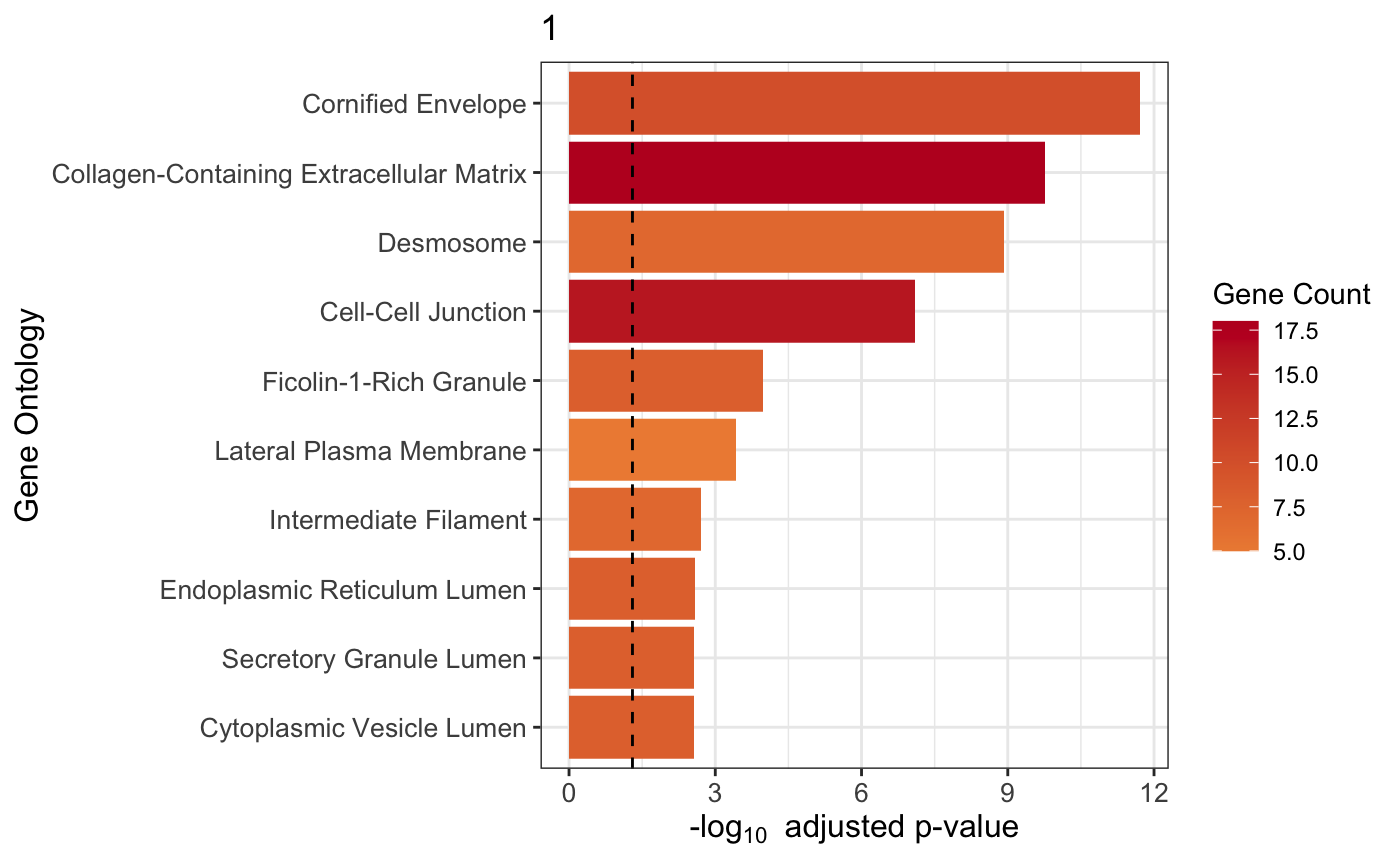
7.2 所有cluster的GO分析
再试试绘制所有cluster的GO分析。🐵
plot_all_cluster_go(markers, org = "human", ont = "BP")

8GSEA分析
8.1 差异分析
首先我们要用find_diff_genes做一下差异分析,寻找差异基因。😜
然后使用test_GSEA()进行GSEA分析。
de <- find_diff_genes(dataset = scRNA_int,
clusters = as.character(0:7),
comparison = c("group", "CTCL", "Normal"),
logfc.threshold = 0, # threshold of 0 is used for GSEA
min.cells.group = 1) # To include clusters with only 1 cell
gsea_res <- test_GSEA(de,
pathway = pathways.hallmark)

8.2 GSEA结果可视化
plot_GSEA(gsea_res, p_cutoff = 0.1, colors = c("#0570b0", "grey", "#d7301f"))

9补充一下
在Scillus包中有一个非常好用的功能叫Plotting Measures,在这里补充一下。😜
在设置的参数中measures = 你需要的基因或指标就可以啦。🧐
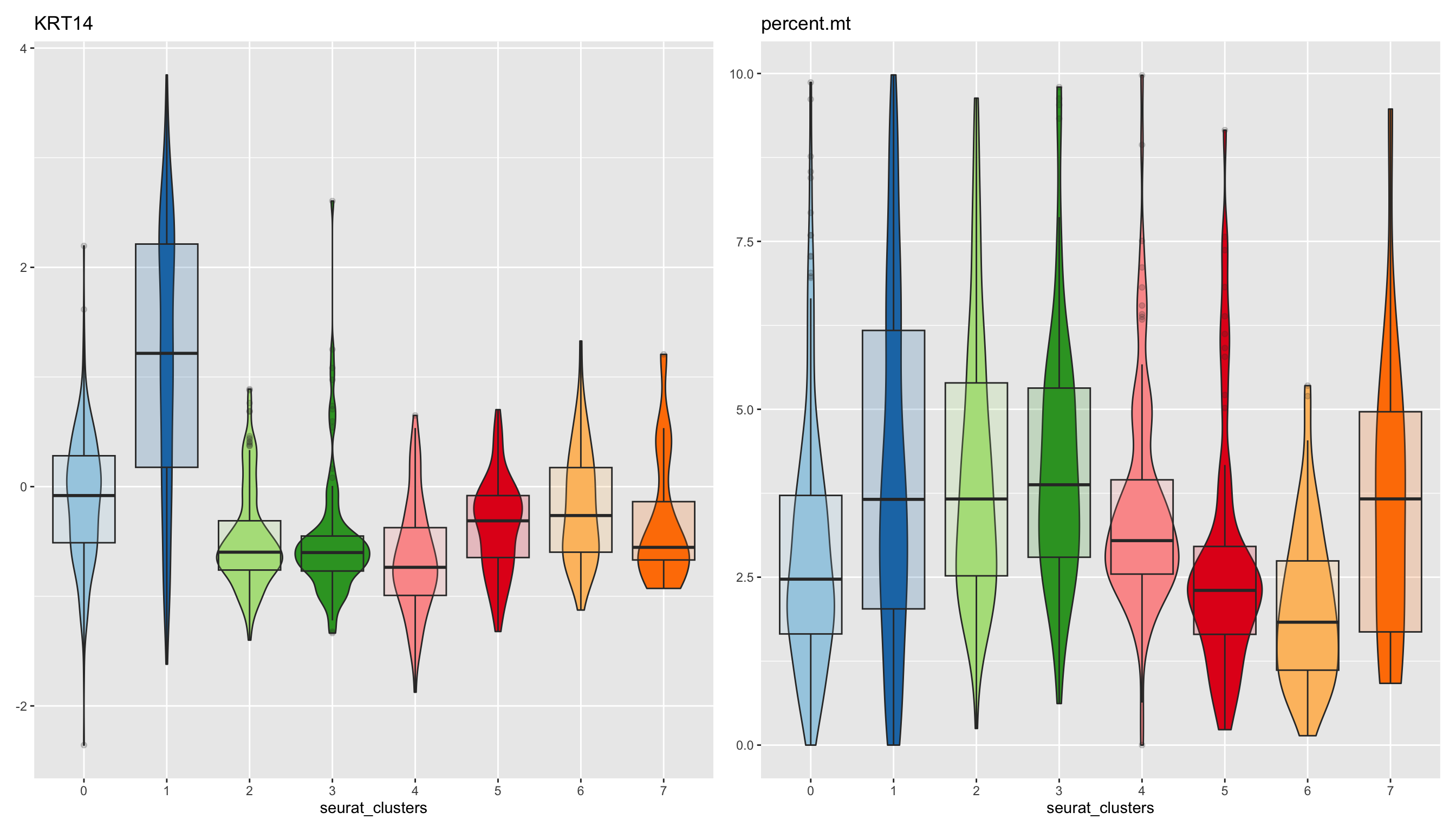
9.1 小试牛刀
plot_measure(dataset = scRNA_int,
measures = c("KRT14","percent.mt"),
group_by = "seurat_clusters",
pal_setup = pal)
9.2 更进一步
plot_measure_dim(dataset = scRNA_int,
measures = c("nFeature_RNA","nCount_RNA","percent.mt","KRT14"),
split_by = "sample")


点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 🤩 WGCNA | 值得你深入学习的生信分析方法!~
📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码!
📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程
📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~
📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~
📍 🧐 rms | 批量完成你的线性回归
📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图
📍 🤠 Network | 高颜值动态网络可视化工具
📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图
📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程)
📍 ......
本文由 mdnice 多平台发布