1写在前面
太久没更了,真是累到极致,每天回到家都只想睡觉。😭
今天介绍一下Scillus包,是一个基于Seurat和ggplot2的R包,用于增强scRNA-seq数据的处理和可视化。🧐
它可以对Seurat对象进行多种类型的图形展示,如热图、聚类图、基因表达图等。🤩
2用到的包
rm(list = ls())
library(tidyverse)
library(Scillus)
library(Seurat)
library(magrittr)
library(purrr)
3示例数据
今天我们用到的是GEO上的公开数据,GSE128531,研究的是皮肤T细胞淋巴瘤(CTCL)的异质性,包含了5名晚期CTCL患者和4名健康捐赠者皮肤活检的14,056个CD3+淋巴细胞(448个细胞来自正常细胞,13,608个细胞来自CTCL皮肤样本)。🥳
原作者也是靠这个scRNA-seq发表了Clin Cancer Res:👇

🔗Data链接在这里:👇
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128531
4环境配置
4.1 文件路径
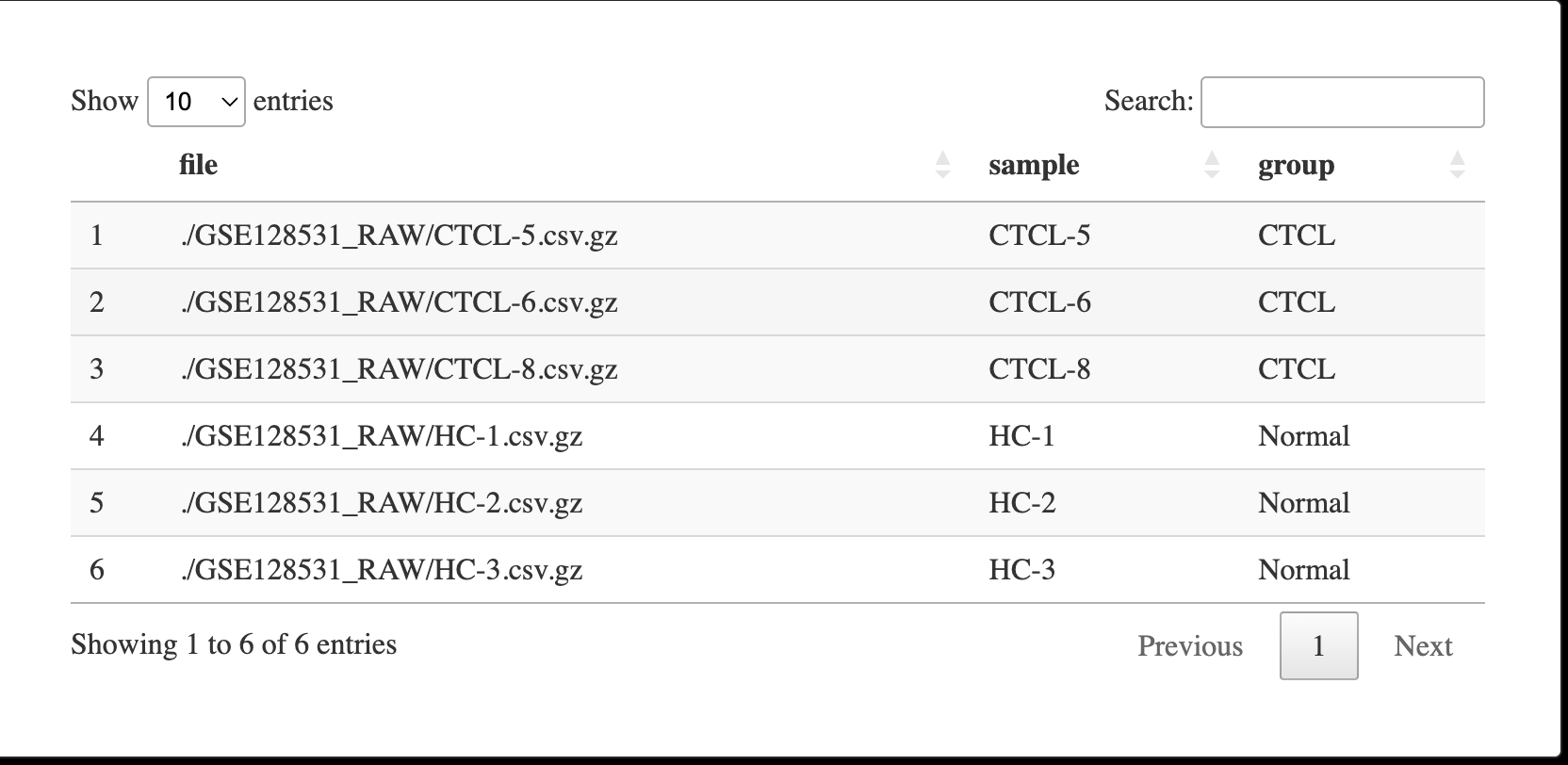
a <- list.files("./GSE128531_RAW", full.names = TRUE)
m <- tibble(file = a,
sample = stringr::str_remove(basename(a), ".csv.gz"),
group = rep(c("CTCL", "Normal"), each = 3))
DT::datatable(m)
4.2 配色
pal <- tibble(var = c("sample", "group","seurat_clusters"),
pal = c("Set2","Set1","Paired"))
4.3 读入数据
这里我们只做3 vs 3的数据处理和可视化哦。🥳
Scillus将为每个样本创建Seurat对象,并自动调用PercentageFeatureSet() 函数来计算线粒体基因的含量。😘
这里,scRNA是一个由多个Seurat对象组成的list。🤓
scRNA <- load_scfile(m)
map(scRNA, print)
4.4 数据长度
这里的长度等于metadata的行数,即m的行数。🤨
length(scRNA)

5QC可视化
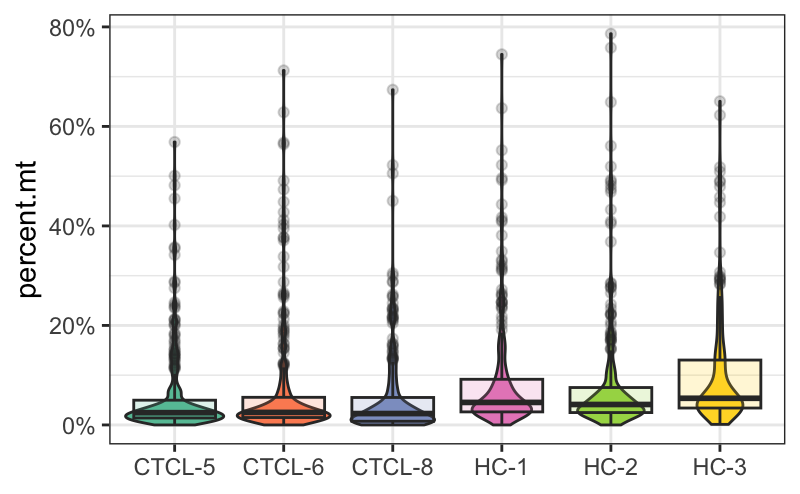
5.1 线粒体基因可视化
plot_qc(scRNA, metrics = "percent.mt")
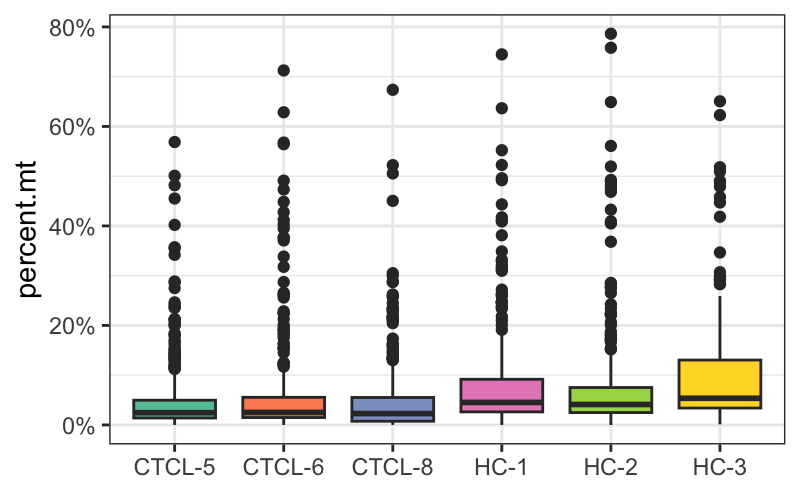
只画boxplot试试。😘
plot_qc(scRNA,
metrics = "percent.mt",
plot_type = "box" # "combined", "box" or "violin"
)
分组比较试试。🐵
默认以sample进行分组,你也可以挑选metadata中的其他列做为分组条件。🍐
plot_qc(scRNA,
metrics = "percent.mt",
group_by = "group" # "sample", 其他在metadata中的列
)

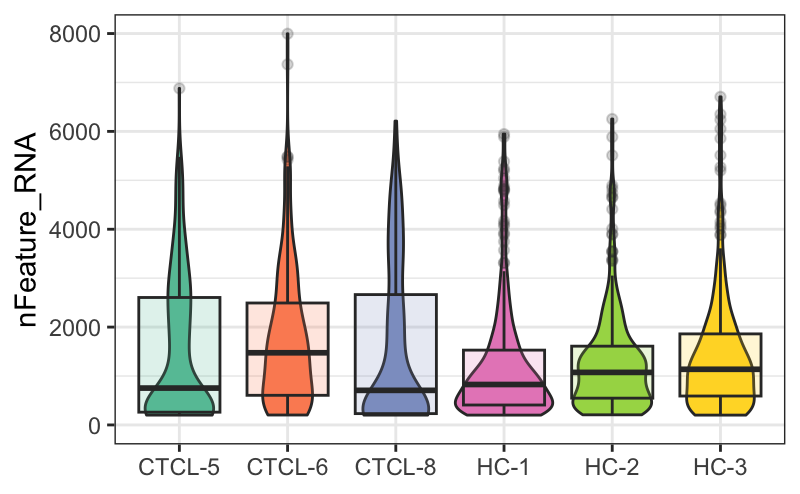
5.2 nFeature可视化
plot_qc(scRNA, metrics = "nFeature_RNA")
换个配色试试。😘
plot_qc(scRNA,
metrics = "nFeature_RNA",
group_by = "group",
pal_setup = "Accent"
)
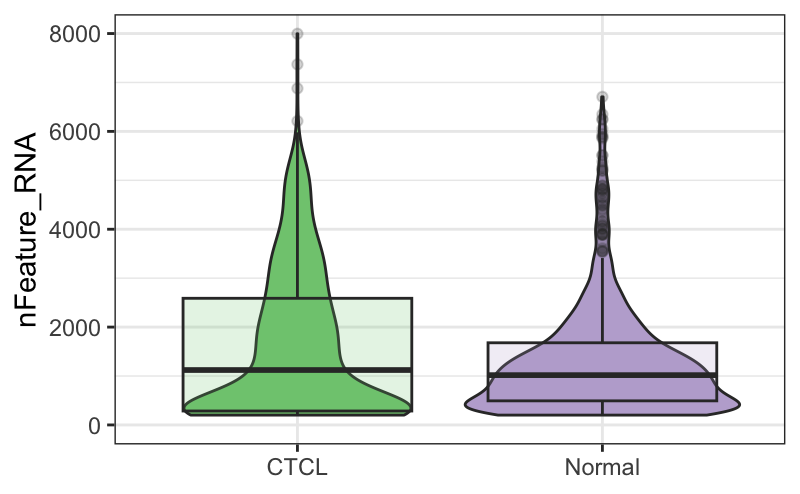
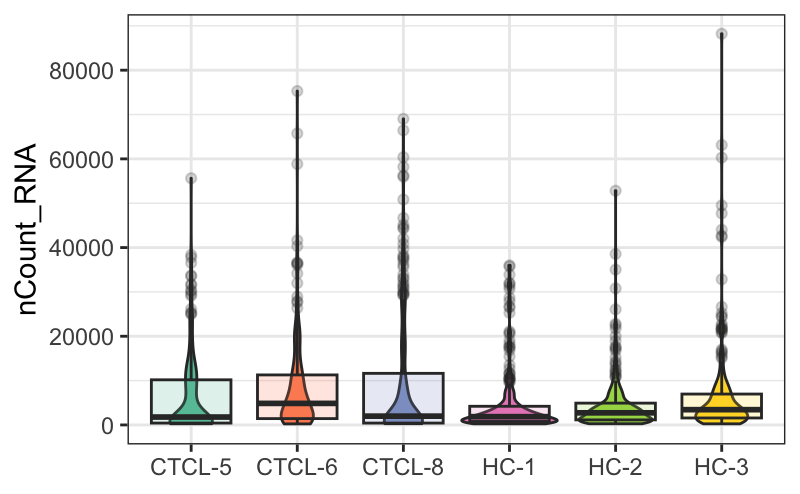
5.3 nCount可视化
plot_qc(scRNA, metrics = "nCount_RNA")
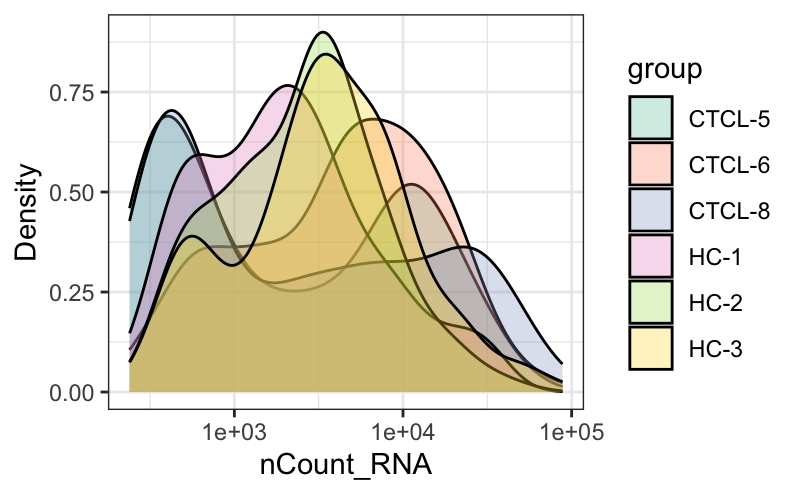
换成density plot试试。😉
plot_qc(scRNA,
metrics = "nCount_RNA",
plot_type = "density") +
scale_x_log10()
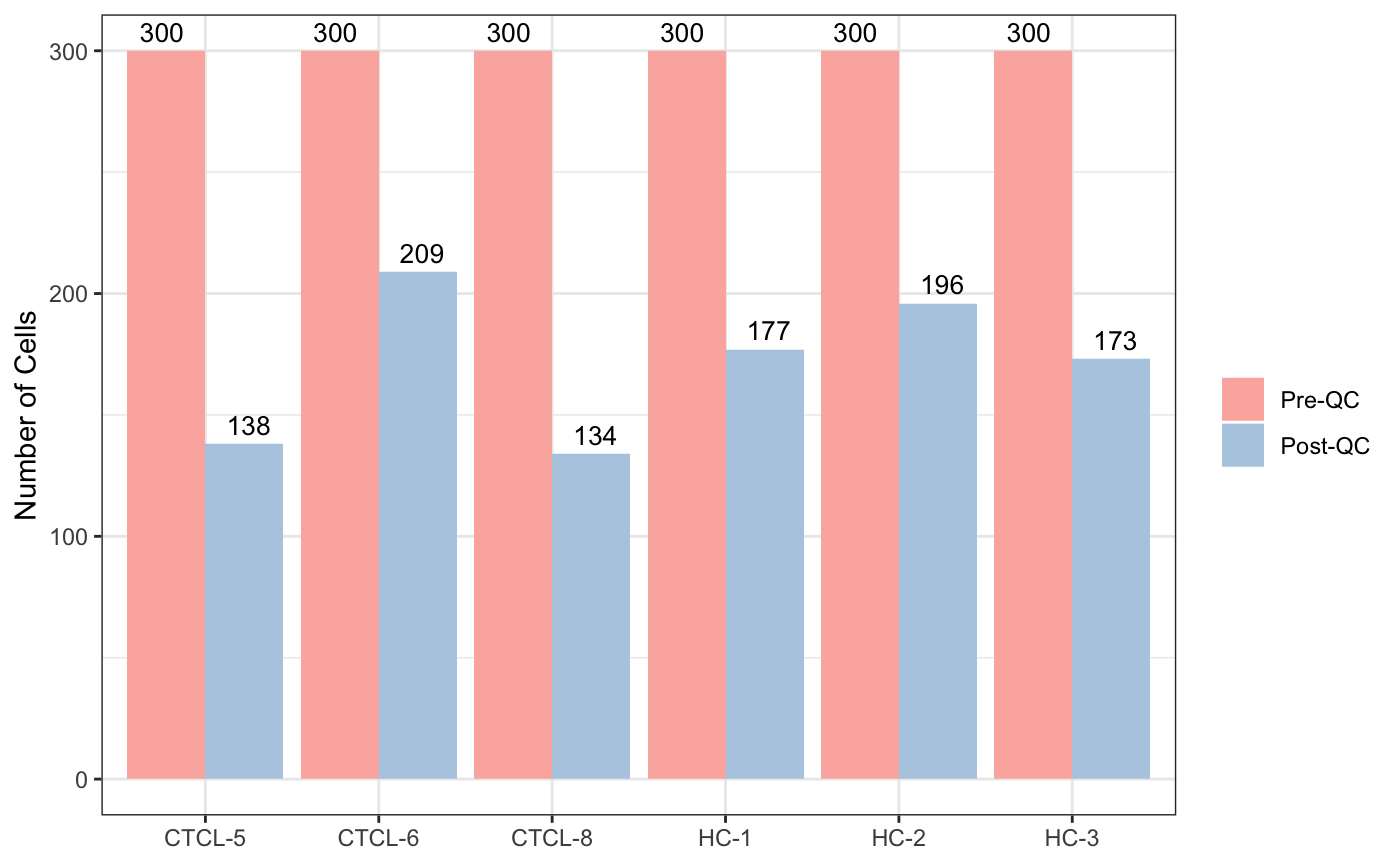
6过滤与整合
6.1 过滤
subset参数的语法与Seurat对象的subset()函数是一样的。🤒
用的时候,会自动绘制barplot以显示过滤前后的细胞数。😉
scRNA_f <- filter_scdata(scRNA, subset = nFeature_RNA > 500 & percent.mt < 10)
6.2 标准化处理
接着就是做一下Normalize,FindVariableFeatures,CellCycleScoring等标准化处理了。🧐
scRNA_f %<>%
purrr::map(.f = NormalizeData) %>%
purrr::map(.f = FindVariableFeatures) %>%
purrr::map(.f = CellCycleScoring,
s.features = cc.genes$s.genes,
g2m.features = cc.genes$g2m.genes)
6.3 整合数据
scRNA_int <- IntegrateData(anchorset = FindIntegrationAnchors(object.list = scRNA_f,
dims = 1:30, k.filter = 50),
dims = 1:30)
scRNA_int %<>%
ScaleData(vars.to.regress = c("nCount_RNA", "percent.mt", "S.Score", "G2M.Score"))
scRNA_int %<>%
RunPCA(npcs = 50, verbose = T)
scRNA_int %<>%
RunUMAP(reduction = "pca", dims = 1:20, n.neighbors = 30) %>%
FindNeighbors(reduction = "pca", dims = 1:20) %>%
FindClusters(resolution = 0.3)
scRNA_int
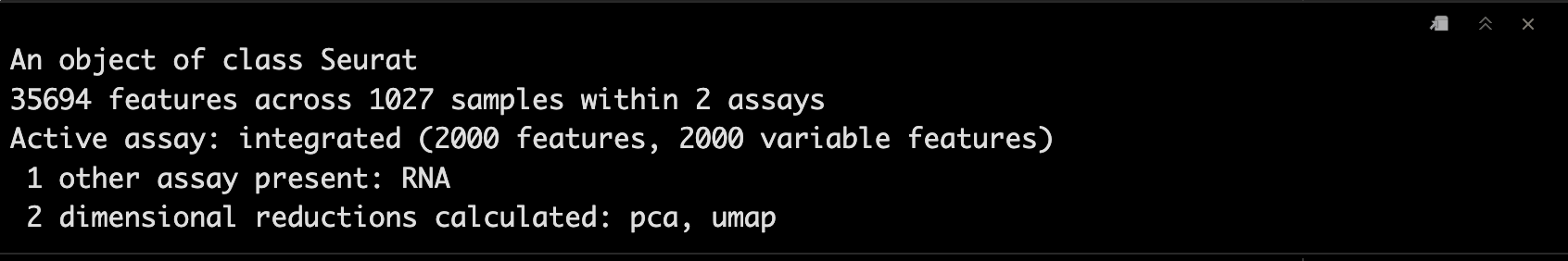
7因子化处理(可选步骤)
主要是处理一下metadata的数据,这样作图会更好看一些,如果你没有metadata,可以不做这一步。😘
m %<>%
mutate(group = factor(group, levels = c("Normal", "CTCL")))
scRNA_int %<>%
refactor_seurat(metadata = m)
scRNA_int

点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 🤩 WGCNA | 值得你深入学习的生信分析方法!~
📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码!
📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程
📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~
📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~
📍 🧐 rms | 批量完成你的线性回归
📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图
📍 🤠 Network | 高颜值动态网络可视化工具
📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图
📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程)
📍 ......
本文由 mdnice 多平台发布