动动发财的小手,点个赞吧!
1. 数据
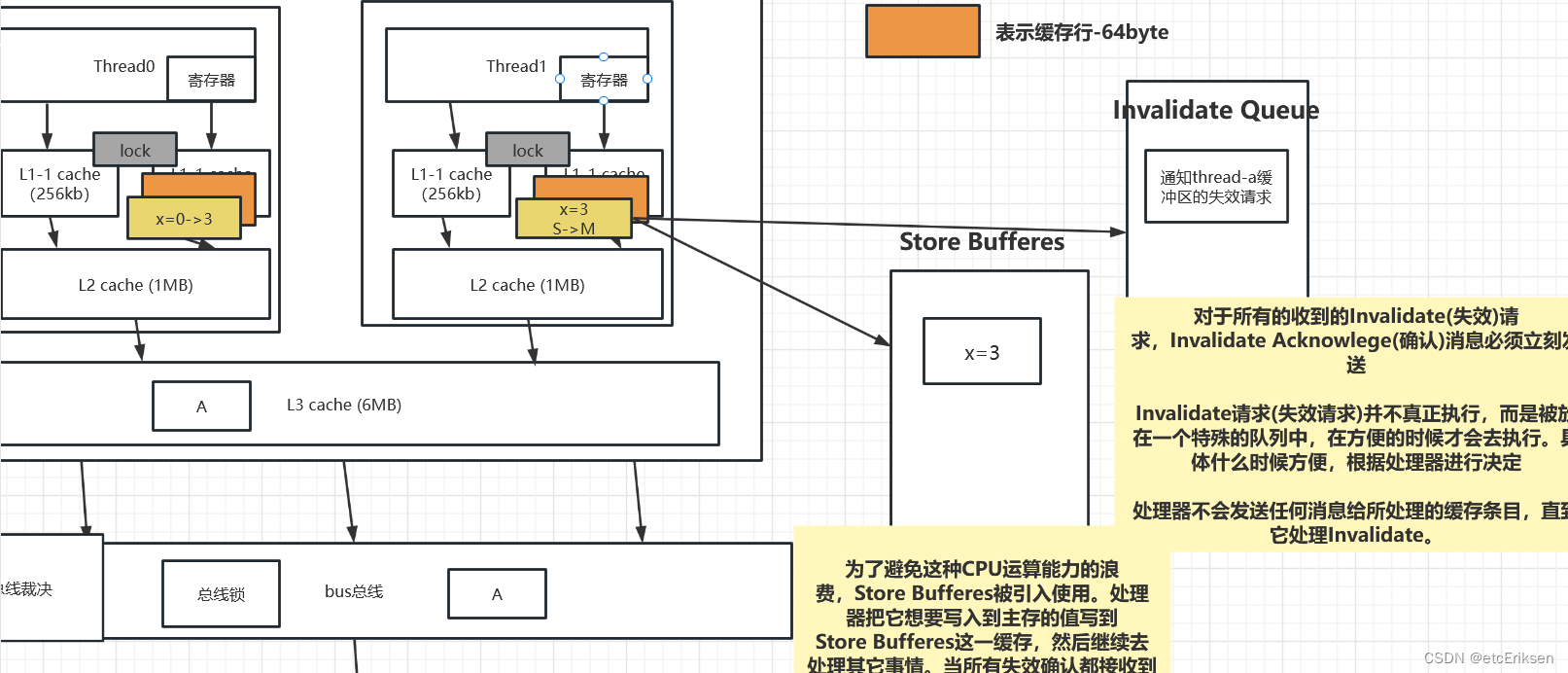
今天,我们将继续回顾我们在上一次中研究的 Myc ChIPseq。这包括用于 MEL 和 Ch12 细胞系的 Myc ChIPseq。
-
可在 此处 [1]找到 MEL 细胞系中 Myc ChIPseq 的信息和文件 -
可在 此处 [2]找到 Ch12 细胞系中 Myc ChIPseq 的信息和文件
在数据目录中,我们按照上一节中概述的处理步骤提供了来自 MACS2 的峰值调用。
MEL 和 Ch12 细胞系中 Myc 的峰值调用可以在:
data/peaks/
-
data/peaks/Mel_1_peaks.xls -
data/peaks/Mel_2_peaks.xls -
data/peaks/Ch12_1_peaks.xls -
data/peaks/Ch12_1_peaks.xls
2. ChIP Peaks
在上一节中,我们回顾了如何使用 MACS2 等峰值调用程序识别假定的转录因子结合位点。
library(GenomicRanges)
macsPeaks <- "data/peaks/Mel_1_peaks.xls"
macsPeaks_DF <- read.delim(macsPeaks,comment.char="#")
macsPeaks_GR <- GRanges(seqnames=macsPeaks_DF[,"chr"],
IRanges(macsPeaks_DF[,"start"],macsPeaks_DF[,"end"]))
mcols(macsPeaks_GR) <- macsPeaks_DF[,c("abs_summit", "fold_enrichment")]
macsPeaks_GR[1:5,]

3. 基因注释
由于转录因子,如名称所示,可能调节其靶基因的转录,我们使用 ChIPseeker 包将代表潜在转录因子结合事件的峰与其重叠或最接近的 mm10 基因相关联。
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
library(ChIPseeker)
peakAnno <- annotatePeak(macsPeaks_GR, tssRegion=c(-1000, 1000),
TxDb=TxDb.Mmusculus.UCSC.mm10.knownGene,
annoDb="org.Mm.eg.db")
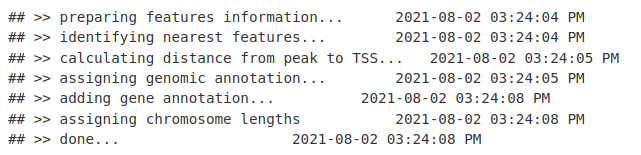
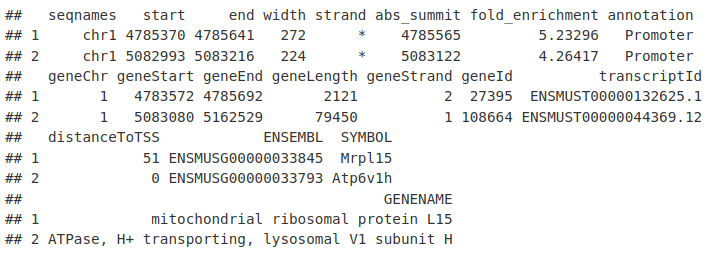
这使我们能够生成峰及其预测目标基因的 GRanges 或数据框。
annotatedPeaksGR <- as.GRanges(peakAnno)
annotatedPeaksDF <- as.data.frame(peakAnno)
annotatedPeaksDF[1:2, ]
参考资料
Data1: https://www.encodeproject.org/experiments/ENCSR000EUA/
[2]Data2: https://www.encodeproject.org/experiments/ENCSR000ERN/
本文由 mdnice 多平台发布