题目:Benchmarking AlphaFold2 on peptide structureprediction
文献来源:2023, Structure 31, 1–9
代码:基准实验,比较了比较多的模型
1.背景介绍
由2-50个氨基酸构成的聚合物可以称为肽。但是关于肽和蛋白质之间的差异还是很模糊。一般来说,除了组成氨基酸的个数,二级结构和三级结构的数量也会影响一个氨基酸到底是属于肽还是蛋白质。一般来说,较少的二级,三级结构的氨基酸链为肽,反之则为蛋白质。肽具有非常灵活的结构,在不同的结合物,不同的时间尺度,甚至与脂质或者蛋白质产生不同的相互作用时,其构象都会不同。目前都是通过NMR的方式来测定液态/固态条件下的小肽结构。但是该方式会因为环境的变化导致预测偏差。而计算方法可以预测肽的稳定构象(可能会具有潜在生物活性),但是这类方法的一致性会因为肽的长度的增加而减少。因此,作者使用AlphaFold2(AF2)对6个不同组的588个肽进行了基准测试,以确定计算方法预测肽结构这类方法的实用性和缺点。
进行多肽结构预测的计算方法包括了从头折叠(de novo folding)、同源建模( homology modeling)、分子动力学(MD)模拟和基于深度学习的方法。PEP-FOLD3是适用于5-50个肽的从头折叠的方法。而APPTEST则是适用于5-40氨基酸的MD+神经网络的方法。AF2是一种基于深度学习的蛋白质预测方法,它使用多序列比对(MSAs),基于共同进化的残基来预测蛋白质的结构。RoseTTAFold通过类似的逻辑,但使用不同的框架进行预测蛋白质结构。Omega-Fold是只使用序列而不使用MSA的深度学习方法。Omega-Fold比较适合没有MSA同源信息的情况下进行预测蛋白质。除了这些方法外,当有同源肽或蛋白质结构可用时,同源建模方法可以进行实验约束的有无或使用野生型实验数据建模蛋白质突变结构。虽然这种方法通常更适用于较大的蛋白质,但它也可以用于肽结构的应用。
AF2的使用是迈向高精度预测蛋白质结构的一大步。理论上来说AF2可以使用NMR实验检测的短一点的肽进行训练建模,但是事实上AF2并没有那么做:它将NMR测出来的短肽数据排除在外了。AF2在CASP14中做出的一些相对较差的预测中包括由NMR确定的蛋白质结构,这就提出了一个问题,即在灵活的肽结构是否也能观察到类似的现象?
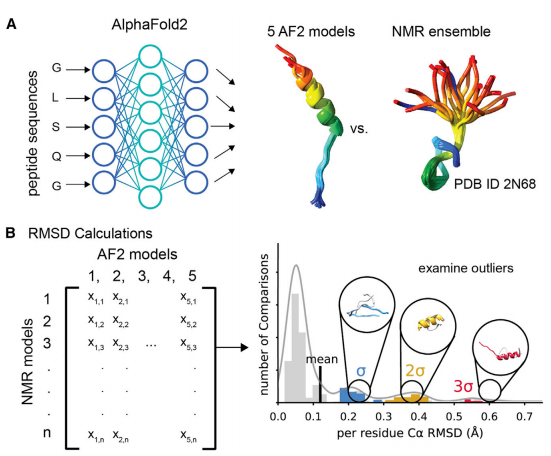
(A)使用AlphaFold2(AF2)预测6个肽类的588个结构,这些结构都已经被NMR方法确定。(B)通过计算CαRMSD来比较AF2针对一个肽序列给出的五个结果和NMR观测出的真实的结构差异。然后将每类肽的RMSD分布绘制成直方图。并且研究了RMSD分别具有一个标准差(σ),两个标准差(2σ)或者三个标准差(3σ),从而了解AF2为什么无法准确预测实验结构。
在这项工作中,作者使用AF2预测10到40肽的结构,并将AF2与其他肽结构预测方法进行比较。首先,他们从PDB24中选择了588个多肽,并使用AF2预测了结构(图1A)。接下来,通过计算预测的结构和实验结构的均方根偏差(RMSD)和Φ/Ψ角恢复来评估的AF2预测性能。作者绘制了RMSD值的分布,以确定AF2预测的离散值。作者通过观察最低RMSD重叠的AF2和NMR结构,逐个地检查了不准确的AF2预测结果,以更好地理解AF2在肽结构预测中的局限性。最后,他们也使用PEPFOLD3、Omega-Fold、RoseTTAFold和APPTEST对588个肽模型进行预测,并与AF2进行统计学比较,且每种方法的效果评价。
2.结果
2.1 结构选择及统计分析
作者选择了588个多肽。这些多肽都具有实验确定的NMR结构,包括明确定义的二级结构元件和无序区域。肽被分为以下几组:螺旋膜相关肽(AH MP)、螺旋可溶性肽(AH SL)、混合二级结构膜相关肽(MIX MP)、混合二级结构可溶性肽(MIX SL)、b发夹肽(BHPIN)和富二硫肽(DSRP)。每种肽的NMR结构的集合都会与AF2给出的5种可能结构进行两两比较,然后得到所有成对Cα RMSD的分布,以确定离群值,并检查预测不佳的结构(图1B)。Cα RMSD仅考虑了肽的二级结构区域,并将其归一化为该区域内的残基数,以避免不同肽的大小变化引起的偏差。

图2 AF2对α-螺旋肽的预测比对膜相关肽的预测表现更好。(A)用于所有NMR和AF2预测归一化的α-螺旋膜肽(AH MP)构象之间比较的Cα RMSD的直方图。平均值和中位数用黑色表示。采用核密度估计方法对数据进行多模态高斯分布拟合。高于平均值的一、二和三个标准差分别用蓝色、黄色和红色表示。(B)AF2预测的RMSD高于平均值的1、2和3个标准差的三个结果。NMR构象以浅灰色表示,AF2的结果以蓝色(一个标准差)、黄色(两个标准差)或红色(接近三个标准差)表示。为了清晰起见,图中提供了PDB ID和规范化的RMSD值。(C)用于所有NMR和AF2预测归一化的α-螺旋可溶性肽(AH SL)构象之间比较的Cα RMSD的直方图。平均值和中位数用黑色表示。采用核密度估计方法对数据进行多模态高斯分布拟合。高于平均值的1、2和三个标准差分别用蓝色、黄色和红色表示。(D)示例模型描述了高于平均值的一、二和三个标准差。每个构象都显示了最低的成对RMSD。NMR构象以浅灰色表示,AF2构象用以蓝色(一个标准差)、黄色(两个标准差)或红色(接近三个标准差)表示。注意,这里的1AMB RMSD略低于3标准差,但这已经代表AH SL数据中最大的离群值。
2.2 AF2在α-螺旋膜相关肽的预测上具有良好的准确性和很少的异常值
这些肽在一个膜环境中会折叠成一个主要的α-螺旋结构。这组肽中主要包括了跨膜螺旋,两亲螺旋,具有螺旋-旋转螺旋基序,以及部分跨膜的单体螺旋等结构。这组数据是数据中的第二大组,由187个肽组成。归一化Cα RMSDs的直方图显示,AF2和NMR模型之间的所有成对比较均为单峰高斯分布,每个残差的均值为0.098A˚(图2A)。作者基于高于平均值的标准差数(σ)来检查了单个离散值,以理解AF2预测的结构缺陷。在某些情况下,AF2不能预测α-螺旋肽的螺旋末端和螺旋-螺旋的末端(图2B)。虽然AF2通过Cα RMSD预测良好,但未能恢复Φ/Ψ角,特别是在低Cα RMSD对的情况下。
2.3 AF2在α-螺旋可溶性肽(AH SL)的预测上显示离散值,并且比在膜相关肽上表现更差
AH SL也是一种α-螺旋肽,其结构在膜环境中没有被识别,在原始出版物中也没有关于膜相互作用的信息,但满足先前描述的a-螺旋膜相关肽的剩余二级结构条件。目前这个组含有41个多肽。归一化Cα RMSD的分布呈双峰高斯分布,每个残差的均值为0.119A˚,第二个峰值在比均值高出2σ到3σ之间(图2C)。图2D显示AF2很难正确预测AH SL的结构。此外,AF2根本不能预测AMB的α-螺旋结构,每个残基的RMSD为0.369A˚(图2D)。最后,AF2也未能恢复AH SL肽的Φ/Ψ角,表明AF2缺乏α-螺旋的想象力。
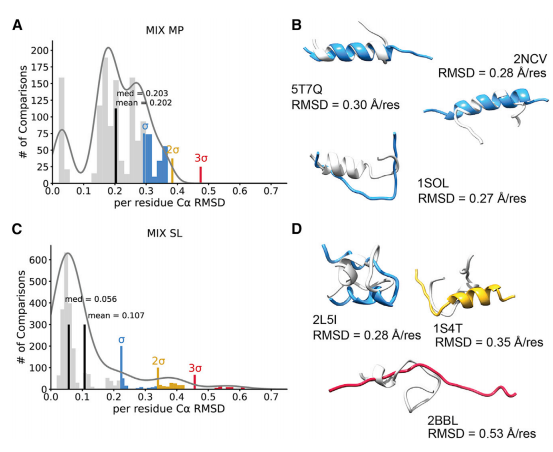
图3 AlphaFold2不能准确预测混合膜肽(MIX MP)和混合可溶性肽(MIX SL)。(A)用于所有NMR和AF2预测归一化的MIX MP构象之间比较的Cα RMSD的直方图。平均值和中位数用黑色表示。采用核密度估计方法对数据进行多模态高斯分布拟合。高于平均值的一、二和三个标准差分别用蓝色、黄色和红色表示。有趣的是,没有一个预测结果低于这一肽类的2σ。(B)AF2预测的三个例子结果显示RMSD的一个标准差高于平均值。NMR模型以浅灰色表示,AF2模型以蓝色表示,为了清晰起见,图中提供了PDB ID和规范化的RMSD值。(C)用于所有NMR和AF2预测归一化的MIX SL构象之间比较的Cα RMSD的直方图。平均值和中位数用黑色表示。采用核密度估计方法对数据进行多模态高斯分布拟合。高于平均值的一、二和三个标准差分别用蓝色、黄色和红色表示。(D)结构例子描述了预测高于平均值的一、二和三个标准差。每个构象都显示了最低的成对RMSD。NMR构象用浅灰色表示,AF2构象用蓝色(一个标准差)、黄色(两个标准差)或红色(三个标准差)表示。
2.4 混合二级结构膜相关肽(MIX MP)在所有数据组中的分布变化最大和RMSD值最大
MIX MP与α-螺旋膜肽等膜相互作用,但它们包含多个一个二级结构区域(例如,由一个旋转分隔的多个α-螺旋、α/β或α/线团混合二级结构等)。这个数据组有14个肽。图3A的归一化的Cα RMSD直方图显示为多个结构的分布,平均每个残基为0.202A˚。AF2预测的结果与相应的NMR预测的结果的偏离均值没有超过2σ,但是整个分布偏向均值,说明其预测较差。图 3B表明,AF2正确地预测了二级结构,但没有与肽段结构较少的区域重叠(图3B)。另一方面,AF2未能预测1SOL的任何二级结构(图3B)。与α-螺旋肽相比,AF2恢复了MIX MP的Φ/Ψ角。
2.5 AF2对混合二级结构可溶性肽具有中等的准确性
混合二级结构可溶性肽(MIX SL)被定义为具有与对应的膜相同二级结构性质的肽,但是这种肽在膜环境中的结构未被识别。这个数据组有21个肽,归一化Cα RMSD直方图显示中等多模态高斯分布,峰值在1σ、2σ和3σ,高于每个残基0.107A˚的平均值(图3C)。离散值表明AF2不能预测二级结构-非结构化边界的方向(图3D)。例如,AF2预测的2BBL是一个完全非结构化的肽,尽管NMR结构在整个集合中包含了一个良好的紧凑结构(图3D)。同样,与α-螺旋肽相比,AF2的Φ/Ψ角RMSD与MH SL肽的Cα RMSD有很好的相关性。
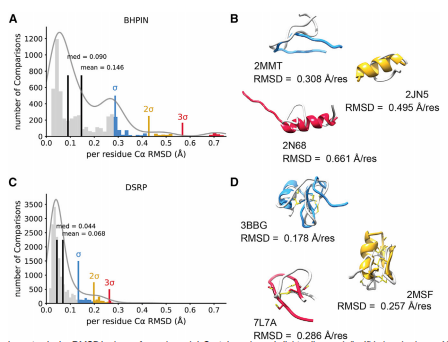
图4 AlphaFold2对b发夹(BHPIN)和富二硫肽(DSRP)的预测。(A)用于所有NMR和AF2预测归一化的BHPIN构象之间比较的Cα RMSD的直方图。平均值和中位数用黑色表示。采用核密度估计方法对数据进行多模态高斯分布拟合。高于平均值的一、二和三个标准差分别用蓝色、黄色和红色表示。(B)结果显示AF2预测的RMSD高于BHPIN类平均值的一个标准差。NMR构象以浅灰色表示,AF2的结果以蓝色(一个标准差)、黄色(两个标准差)或红色(接近三个标准差)表示。为了清晰起见,图中提供了PDB ID和规范化的RMSD值。(A)用于所有NMR和AF2预测归一化的DSRP构象之间比较的Cα RMSD的直方图。平均值和中位数用黑色表示。采用核密度估计方法对数据进行多模态高斯分布拟合。高于平均值的一、二和三个标准差分别用蓝色、黄色和红色表示。(D)结构例子描述了预测高于平均值的一、二和三个标准差。每个模型都显示了最低的成对RMSD。NMR模型用浅灰色表示,AF2模型用蓝色(一个标准差)、黄色(两个标准差)或红色(三个标准差)表示。半胱氨酸显示为浅黄色和二硫键显示。
2.6 AF2对b-发夹(BHPIN)肽(无论是吻合肽还是非吻合肽)都有良好的准确性
b发夹肽组包括具有单一b发夹基序的肽组。这个组的成员可以通过二硫键的存在来固定,也可以不固定。这组共59个肽(26个用二硫键连接,33个未连接)。总的来说,AF2在BHPIN肽上表现良好。AF2和NMR模型之间的所有成对Ca RMSD比较的直方图显示了一个双峰分布,峰值高于每个残基的平均值0.146A˚1σ(图4A)。高于平均值的AF2的把一些BHPIN结构预测为螺旋形,导致较差的RMSD值(图4B)。有趣的是,所有高于平均值的模型都没有连接。对于BHPIN,Φ/Ψ角恢复预测优于在混合二级结构肽的表现,但低于α-螺旋肽(图S3A),表明AF2的β-片Φ/Ψ角想象力优于α-螺旋想象力。
2.7 富二硫肽(DSRP)结构预测具有很高的准确性,但会随着二硫键模式变化
富二硫键肽(DSRP)在本工作中被定义为有任何两个或两个以上二硫键的肽。这一组包括毒素肽,如α-锥形毒素,由多个二硫键环化的b-发夹,以及一些激素肽。DSRPs是中最大的数据组,共包含266个肽。DSRPs显示了一个紧密的、略带双峰的高斯直方图,峰值比每个残差的均值0.068A˚高出两个标准差(图4C),这是所有组中最低的平均值。异常值未能预测出正确的二硫键模式。3BBG正确预测一个,并将大多数剩余的半胱氨酸残基放置在附近,2MSF错位了两个二硫化物,未能预测另一个的键,7L7A无法预测任何二硫键(图4D)。与BHPIN一样,DSRPs恢复的Φ/Ψ角角度优于混合角度,但比α-螺旋角度差。有趣的是,AF2未能预测25个DSRPs正确的二硫键。这些肽经常包含连续的半胱氨酸,或预测的AF2结构会显示二硫键的损失。奇怪的是,通过归一化Ca RMSD,25个中只有10个是统计异常值,说明AF2预测结构虽然正确,但仍然缺少正确的二硫键构象。
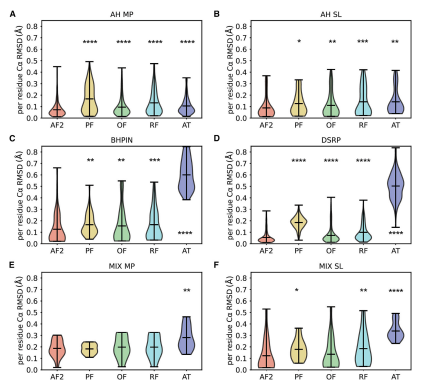
图5 AlphaFold2比其他计算方法、Omega-Fold(OF)、PEPFOLD-3(PF)、RoseTTAFold(RF)和 APPTEST(AT)更能预测肽结构。(A-F)每种计算预测方法和NMR的二级结构区域之间的比较的平均α RMSD的分布。肽组(A)螺旋膜蛋白(AH MP)、(B)螺旋可溶性肽(AH SL)、(C)b发夹肽(BHPIN)、(D)富二硫肽(DSRP)、(E)混合膜肽(MIX MP)和(F)混合可溶性肽(MIX SL)。除MIX MP和MIX SL外,在所有肽类中,AlphaFold2的表现在统计学意义上显著优于PEPFOLD3、Omega-Fold、RoseTTFold和appt。对于MIX MP,除应用测试外,所有方法都具有相似的精度。有趣的是,Omega-Fold对于MIX SL的表现和AF2一样好,而且两者都优于其他方法。采用配对双侧t检验计算各分布对AF2和*p < 0.05、**p < 0.01、****p<0.001和****p < 0.0001值的统计差异。
2.8 多肽计算结构预测方法的统计评价
作者试图了解AF2在预测肽的实验结构方面是否比其他深度学习和从头蛋白/肽预测方法具有优势。他们使用PEPFOLD3、Omega-Fold、RoseTTAFold和apptest对我们所有588个肽的结构进行了预测。PEPFOLD3和APPTEST用于肽结构预测,而Omega-Fold和RoseTTAFold用于一般的蛋白质结构预测。通过长度归一化Cα RMSD测量,AF2比AH MP、AH SL、BHPIN和DSRP更能准确地预测肽结构(图5A-5D)。有趣的是,AF2仅在MIX MP上优于apptest(图5E)。最后,AF2优于PEPFOLD3、RoseTTAFold和APPTEST,但在MIX SL肽方面的表现与Omega-Fold一样好(图5F)。
2.9 预测排名与预测精度之间的相关性较差
一个标准的AF2运行会生成5个结构作为输出。这些结构根据其预测的局部距离差检验(pLDDT)值对程序进行排序,pLDDT是一个估计预测结构与基于主碳坐标的实验结构的一致性的指标。最低排名的结构代表了对给定序列的最佳预测。然而,这个假设可能并不总是正确的。本研究将生成的所有5个AF2结构与相应的NMR结构进行两两比较,以获得更好的结构采样。理论上,较低级阶的结构应该给出最低的Cα RMSD值。为了检验这一假设是否正确,作者列举了每个等级的最低Cα RMSD成对比较。基于这些总和,AF2分配的前三个排名与给出Cα RMSD最低的结构之间没有相关性。然而,排名第四和第五的预测结构在最低的Cα RMSD中较少。因此,研究结果表明,AF2用于评估全局蛋白结构的pLDDT度量并不是评估肽的构象的一个有意义的度量。
2.10 该研究的局限性
该研究的第一个局限性是由于许多肽的内在灵活性。肽可以具有高度灵活的区域,包括线圈或转弯,这可能导致相同结构的多种构象。NMR结构通常由一个构象集合组成,这使得预测结构和实验结构之间的精确比较具有挑战性。为了解决这一点,作者将NMR构象与每种方法的所有预测输出进行两两比较,得到比较的分布,并选择最低的Cα RMSD进行说明和方法之间的统计比较。此外,这些数据的多肽段的NMR和x射线结构测量差异不会导致在AF2的性能方面的显著差异。
另一个与灵活性相关的一点是,在NMR识别的结构中,可替代的低能构象可能存在。特别是对于具有多个通过旋转或线圈连接的结构域的螺旋,实验方法捕获的结构可能只代表多肽的多种构象中的一种。从这个角度来看,AF2预测的结构并不一定是错误的,但它们可能只是对应于肽的另一种构象,尽管AF2预测的x射线结构同样准确。一项旨在比较AF2预测结构与溶液中NMR确定的实验结构的研究发现,在某些情况下,AF2预测的结构比NMR更稳定,因此它的预测可能可行。
最后,一些多肽可能会折叠成不同的二级结构,这种情况取决于环境条件,包括pH值、温度和膜的存在。由于NMR实验的结构是通过一组实验参数获得的,AF2预测可能代表了在不同条件下可以获得的肽的不同构象。因此,有必要研究AF2是否对特定条件下确定的结构是否存在偏见。
3 讨论
除少数异常值外,AF2在α-螺旋膜肽结构预测任务总体上取得了良好的成功。离散值没有显示出造成偏差的单一原因,但AF2不能预测某些肽的二级结构和对扭结角的不准确预测是造成偏差的主要原因。混合二级结构肽的预测也有不同的准确性。在膜相关肽的情况下,偏差主要是由于预测α-螺旋区域之间的角度的误差导致的,但二级结构的预测总体上比较准确,很少有例外。
将螺旋膜蛋白与螺旋溶剂化蛋白进行比较时,AF2对膜溶剂化蛋白的预测表现得更好(RMSD平均为0.092A˚vs0.130A˚)。相比之下,当比较混合膜蛋白和混合溶剂化蛋白时,后者表现更好(RMSD分别为2.17A˚和1.26A˚)。这表明二级结构比溶剂化信息更能影响AF2的精度。然而,AF2在溶剂化较少的BHPIN和DSRP上的性能提高,支持了高溶剂化肽仍然难以预测的观点(图3和4)。溶剂化肽可能根本不需要基于MSA的深度学习模型,因为只需要序列信息的Omega-Fold的性能与AF2在这个肽的结构预测任务上的表现相当。这表明溶剂化肽结构预测很少从MSAs固有的进化数据中获益。
另一方面,b-发夹肽和DSRPs是更紧凑的,更少的溶剂暴露的肽,并且AF2的性能超过了Omega-Fold和该组中的所有其他方法。DSRP在测试肽中具有最高的准确性,这可能是由于存在多个二硫键导致的受限结构产生的影响。这些结构也具有明确的二级结构的区域,与存在多个自由度的螺旋区域相比,AF2预测这种结构会更容易。AF2对DSRPs的预测在统计学上显著优于任何其他的计算方法检验。另一方面,结果表明,在某些情况下,最低的RMSD AF2预测具有不同的二硫键模式或根本没有二硫键。这表明,在预测可能具有替代二硫键模式的DSRP结构时,必须特别小心,并且在AF2运行后,可能需要使用额外的工具来重塑二硫键。
除溶剂化肽外,大多数被测肽组的Φ/Ψ角恢复率较差。考虑到其他研究中AF2的球状蛋白的计算出的转子回收率超过80%的成功现象,作者觉得造成这种差异的原因可能是,由于小的和扩展的支柱,所研究的肽大多具有暴露于溶剂中的残基,很少有埋藏的氨基酸。虽然这可能与AF2无法捕获这些柔性区域的正确几何形状有关,但也有可能NMR结构没有在实验条件下溶解,。作者探索了计算出的Φ/Ψ角百分比与RMSD等参数之间的相关性,但但是只有溶剂暴露肽存在相关性。考虑到全局蛋白中Φ/Ψ角的大偏差,可能需要额外的工具来优化AF2生成的肽的二级结构区域或侧链构象。如 Rosetta二硫化、侧链优化、或简单的势能函数最小化,可能有助于优化初始的AF2模型。
最后,作者的分析表明,pLDDT是AF2用来对生成的结构进行排序的主要指标,但它并不能很好地衡量肽结构是否被准确预测。在与NMR集成的最低RMSD两两比较中,pLDDT的第一、第二和第三级的表现相对一样,而第4和第5级的表现较少(图S5)。一项利用AF2研究蛋白质折叠的多个方面的研究,涉及检测不同长度蛋白质的pLDDT值,结果表明,较短的序列一般往往有较大的pLDDT值。这可能会缩小好和坏预测结构之间的差距,从而降低该指标对小肽结构的作用。在对AF2生成的结构缺乏一个明确的选择标准的情况下,可能有必要增加生成的输出结构的数量,并使用聚类等方法来缩小更频繁地采样的构象或显示更一致的模式来选择准确的结构。
那么这就提出了一个问题:本研究中发现的AF2的缺陷问题是肽特有的,还是与AF2未训练的结构进行预测相关的普遍问题。毫不奇怪,AF2在预测一类肽上存在缺陷,例如被排除在初始训练集中的肽,并表明在未来的训练数据可能会提高这些系统的性能。然而,AF2在高接触肽类别中仍然优于肽特异性方法。奇怪的是,AF2在短肽上的表现优于长肽。自从AF2发布以来,基准研究的重点一直是AF2能够做什么,而很少关注它的缺点。该工作提供了一个建模肽时应该考虑的限制因素。
4 总结
如果预期目标肽具有明确的二级结构,且缺乏具有不同构象的多个旋转或灵活区域,则AF2可用于对小于40个氨基酸的肽结构的建模。AF2在预测a-螺旋膜相关肽和DSRP方面特别成功,但在扩展螺旋或柔性区域的情况下降低了准确性。即使是对于DSRP,问题二硫键模式可能导致肽预测的错误。此外,AF2的预测显示预测的排名和预测精度之间没有相关性,因此可能有必要从AF2生成的结构中选择其他指标。总的来说,使用AF2来预测肽结构需要开发额外的指标和控制,以提高其准确性。
-------------------------------------------
欢迎点赞收藏转发!
下次见!