1. 数据
今天将继续回顾我们在上一次中研究的 Myc ChIPseq。这包括用于 MEL 和 Ch12 细胞系的 Myc ChIPseq 及其输入对照。
-
可在 此处 [1]找到 MEL 细胞系中 Myc ChIPseq 的信息和文件 -
可在 此处 [2]找到 Ch12 细胞系中 Myc ChIPseq 的信息和文件 -
可以在 此处 [3]找到 MEL 细胞系的输入控制 -
可在 此处 [4]找到 Ch12 细胞系的输入对照。
2. 质量控制
ChIPseq 有许多潜在噪声源,包括 * 抗体的不同效率 * 非特异性结合 * 文库复杂性 * ChIP 伪影和背景。
许多这些噪声源都可以使用一些完善的方法进行评估。
2.1. 质控参考
-
Encode 质量指标。
Large-scale quality analysis of published ChIPseq data. Marinov GK, Kundaje A, Park PJ, Wold BJ. G3 (Bethesda). 2014 Feb 19;4(2)
-
ChIPseq 中人工制品重复的高估。
Systematic evaluation of factors influencing ChIPseq fidelity.Nat Methods. Chen Y, Negre N, Li Q, Mieczkowska JO, Slattery M, Liu T, Zhang Y, Kim TK, He HH, Zieba J, Ruan Y, Bickel PJ, Myers RM, Wold BJ, White KP, Lieb JD, Liu XS. 2012 Jun;9(6)
-
什么时候 QC 有用。
Impact of artifact removal on ChIP quality metrics in ChIPseq and ChIP-exo data.Front Genet. 2014 Apr 10;5:75.Carroll TS, Liang Z, Salama R, Stark R, de Santiago I
2.2. 合适的输入
-
在 IP 富集之前,输入样本通常由片段化的 DNA 制成。 -
允许控制样本中出现的伪影区域。 -
切勿在不考虑使用哪个输入的情况下运行 ChIPseq。
例如:当使用肿瘤样本进行 ChIPseq 时,匹配输入样本很重要。同一组织的不同条件可能共享共同的输入。
2.3. 质量指标
ChIPQC 包将一些指标包装到 Bioconductor 包中,并注意在适当的条件下测量这些指标。
要运行单个样本,我们可以使用 ChIPQCsample() 函数、相关的未过滤 BAM 文件,我们建议提供黑名单作为 BED 文件或 GRanges 和基因组名称。
您可以在 Anshul Kundaje[5] 的网站或直接从 Encode[6] 网站找到大多数基因组的黑名单
QCresult <- ChIPQCsample(reads = "/pathTo/myChIPreads.bam", genome = "mm10", blacklist = "/pathTo/mm10_Blacklist.bed")
我们从 Encode 下载 mm10 的黑名单。然后,我们可以使用 ChIPQC 包中的 ChIPQCsample() 函数对我们的 ChIPseq 样本质量进行初步分析。
在这里,我们评估我们在之前的会话中使用 Rsubread 对齐的样本的质量。返回的对象是 ChIPQCsample 对象。
library(ChIPQC)
toBlkList <- "~/Downloads/ENCFF547MET.bed.gz"
chipqc_MycMel_rep1 <- ChIPQCsample("SR_Myc_Mel_rep1.bam", annotation = "mm10", blacklist = toBlkList,
chromosomes = paste0("chr", 1:10))
class(chipqc_MycMel_rep1)

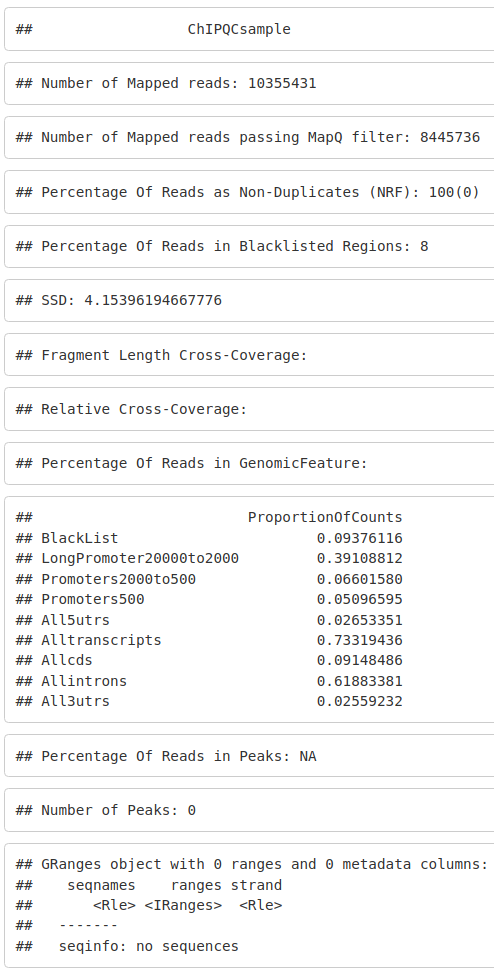
我们可以显示我们的 ChIPQCsample 对象,它将显示我们的 ChIPseq 质量的基本摘要。
chipqc_MycMel_rep1
2.4. 多样本QC
最好对照您的输入对照和我们正在使用的其他 Myc 样本(如果您没有自己的数据,甚至是外部数据)检查 ChIPseq 质量。
这将使我们能够识别样本与对照中 ChIPseq 富集的预期模式,并通过这些指标发现任何异常样本。
我们可以使用 lapply 对所有感兴趣的样本运行 ChIPQCsample()。
bamsToQC <- c("Sorted_Myc_Ch12_1.bam", "Sorted_Myc_Ch12_2.bam", "Sorted_Myc_MEL_1.bam",
"Sorted_Myc_MEL_2.bam", "Sorted_Input_MEL.bam", "Sorted_Input_Ch12.bam")
myQC <- bplapply(bamsToQC, ChIPQCsample, annotation = "mm10", blacklist = toBlkList,
chromosomes = paste0("chr", 1:10))
names(myQC) <- bamsToQC
所有 ChIPQC 函数都可以与 ChIPQCsample 对象的命名列表一起使用,以将分数聚合到表和图中。
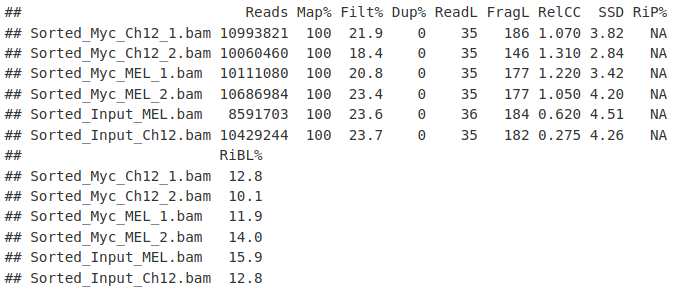
在这里,我们使用 QCmetrics() 函数来概述质量指标。
QCmetrics(myQC)
参考资料
Myc: https://www.encodeproject.org/experiments/ENCSR000EUA/
[2]Ch12: https://www.encodeproject.org/experiments/ENCSR000ERN/
[3]MEL: https://www.encodeproject.org/experiments/ENCSR000ADN/
[4]Ch12: https://www.encodeproject.org/experiments/ENCSR000ERS/
[5]Anshul Kundaje: https://sites.google.com/site/anshulkundaje/projects/blacklists
[6]Encode: https://www.encodeproject.org/annotations/ENCSR636HFF/
本文由 mdnice 多平台发布







![[安装] Dell电脑安装系统时看不到固态硬盘的解决方案](https://img-blog.csdnimg.cn/img_convert/fc95cc2782e24f7aba4f172cc82b5039.png)