TIDE(Tumor Immune Dysfunction and Exclusion) 是一个用于预测癌症患者对免疫检查点抑制剂(如PD-1/PD-L1抑制剂)反应的算法。研究者通过检测肿瘤建模队列中每个基因的表达与效应性毒性T淋巴细胞(CTL)浸润水平的相互关系及对生存情况的影响,从而构建算法来鉴定其他肿瘤队列中的T细胞功能障碍特征。
研究者构建的TIDE算法模拟肿瘤免疫逃逸/抑制的机制主要是两种:
1、CTL功能失调(dysfunction) :在免疫细胞高浸润的肿瘤中,存在着功能失调的效应性毒性T细胞,这些T细胞本可以有效地杀伤肿瘤细胞,但由于某种原因它们的功能被抑制。
2、T细胞清除/排除(exclusion) :在某些肿瘤中,尽管免疫系统可以识别肿瘤,T细胞却无法有效浸润到肿瘤内部,反而被排除在外。
换句话说,该预测工具主要是是通过分析队列中CTL/T细胞的情况进行免疫反应的预测。

该研究团队开发了门户网站和python代码两种分析方式。本次演示网页版的使用方法。
首先进入门户网站: http://tide.dfci.harvard.edu/,在门户网站的中间处可以看到一个视频介绍使用该网站/方法的注意事项。
注意事项主要有以下几点
1、进入界面后需注册后使用
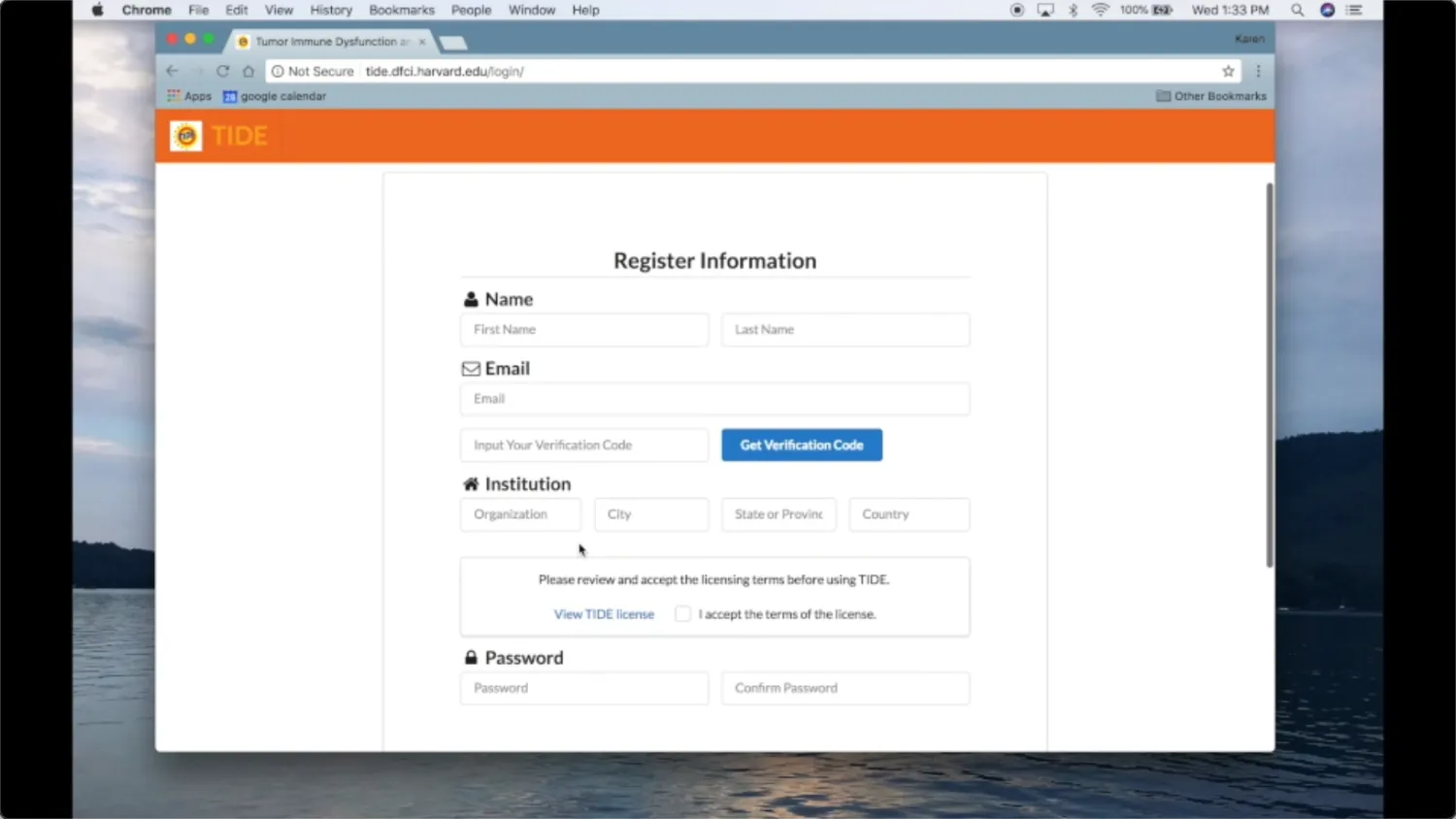
2、需输入的信息
基因表达矩阵、癌症类型和样本数据是否既往接受过免疫治疗

分别对应以下三个红色方框,其中

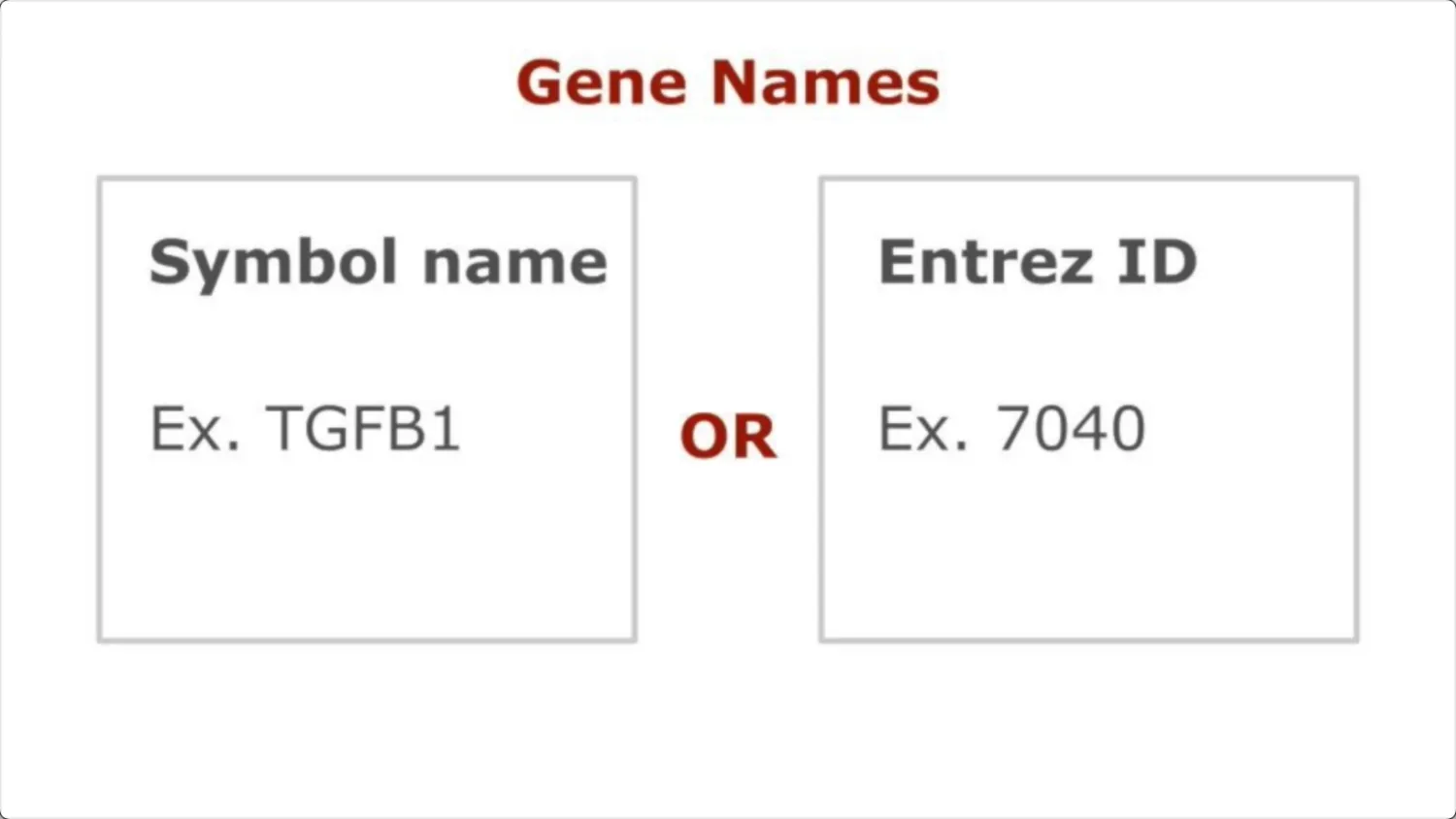
3、表达矩阵格式
应是txt,tsv格式的数据,行是基因数据,列为患者样本信息。行名可以是symbol 或者 Entrez ID。
在网页的下方有示例数据,可以参照修改。

4、表达矩阵信息
需要标准化处理,首选的是用正常样本或者不同肿瘤的混合样本作为参考队列。如果没有参考样本,可以选择用所有样本的平均值作为参考队列标准化处理。
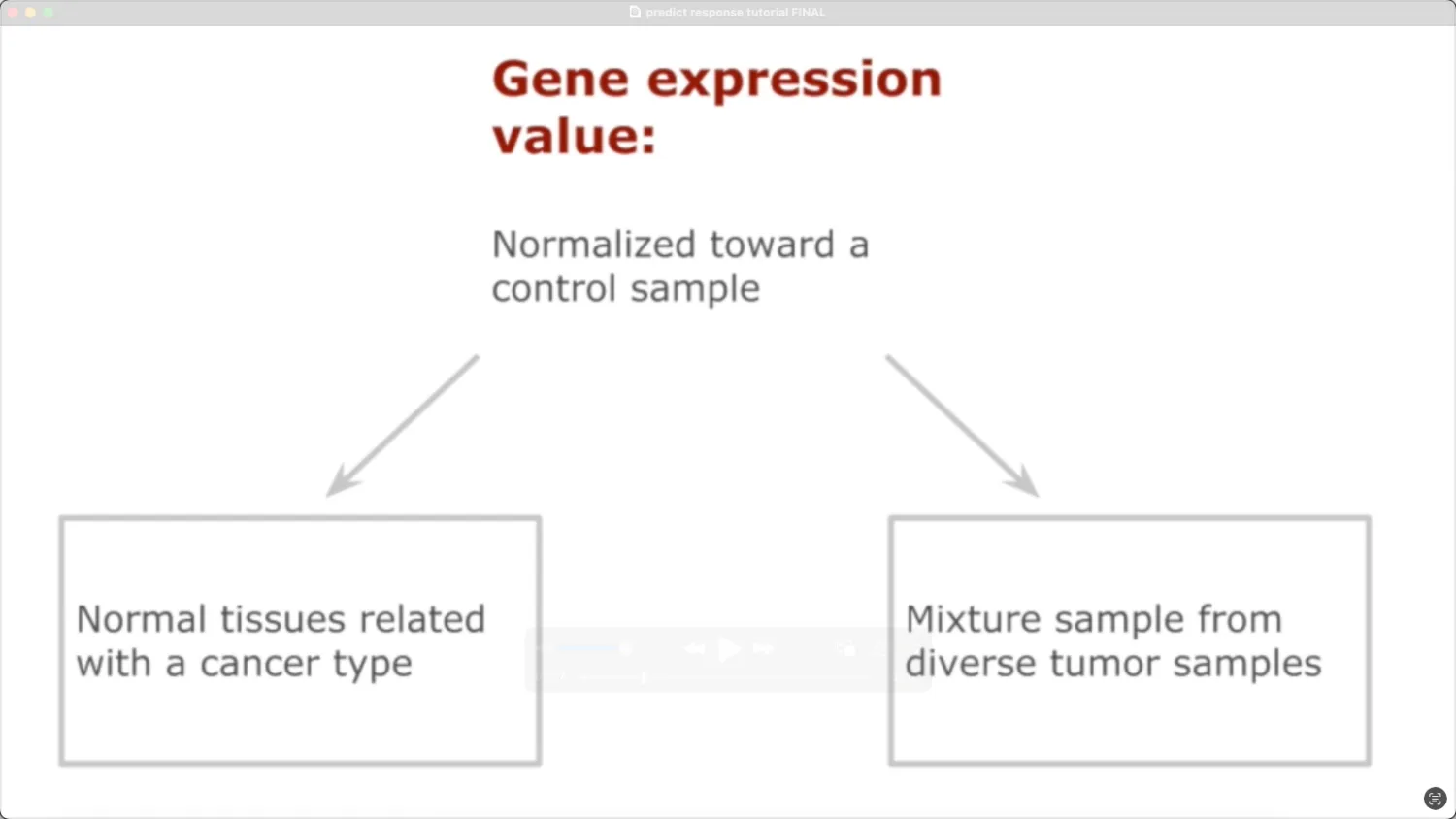

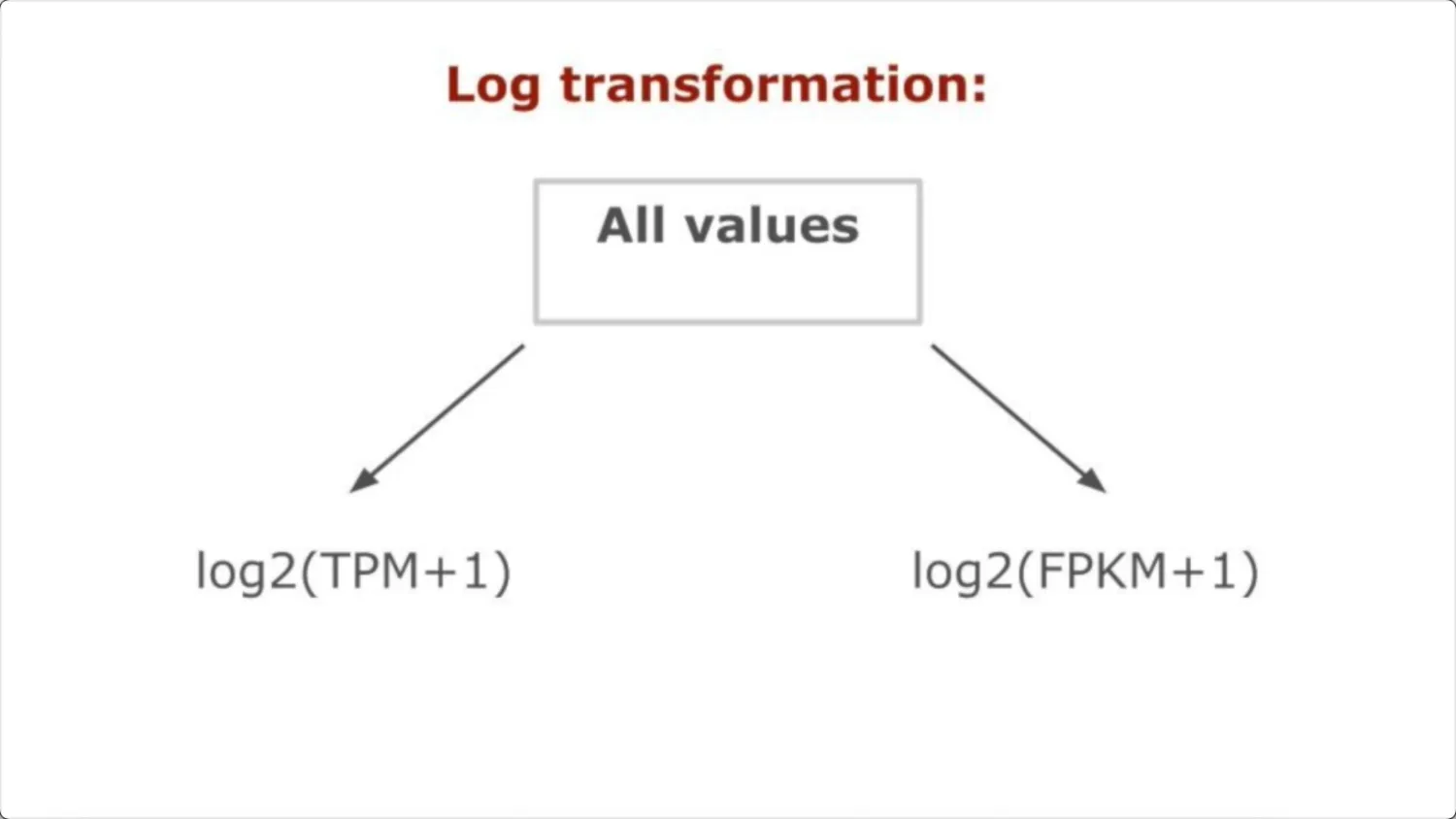
RNA-seq数据应使用log2之后的TPM或者FPKM数据。TPM和FPKM数据不可用于分析,除非有质量良好的参考队列。没有特别注明是否可用或不可用芯片数据。

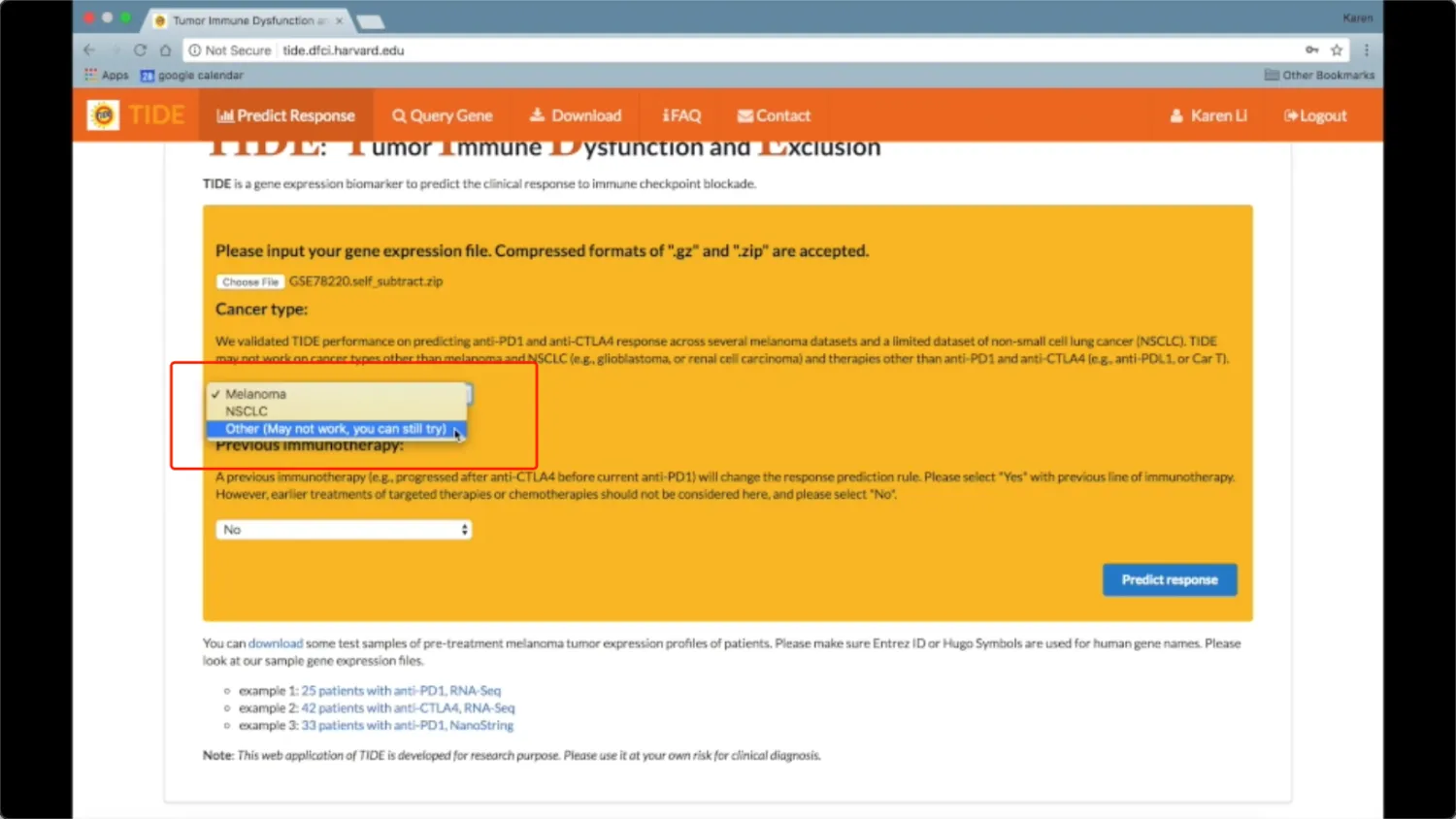
5、癌症类型选择
主要分三种:黑色素瘤,非小细胞肺癌,和其他。由于建模采用的数据集是肿瘤接受PD1和CTLA4治疗反应队列,所以预测的肿瘤也应当是可接受PD-1/CTLA4治疗的。因此,需要提前了解所研究肿瘤目前的治疗方案!
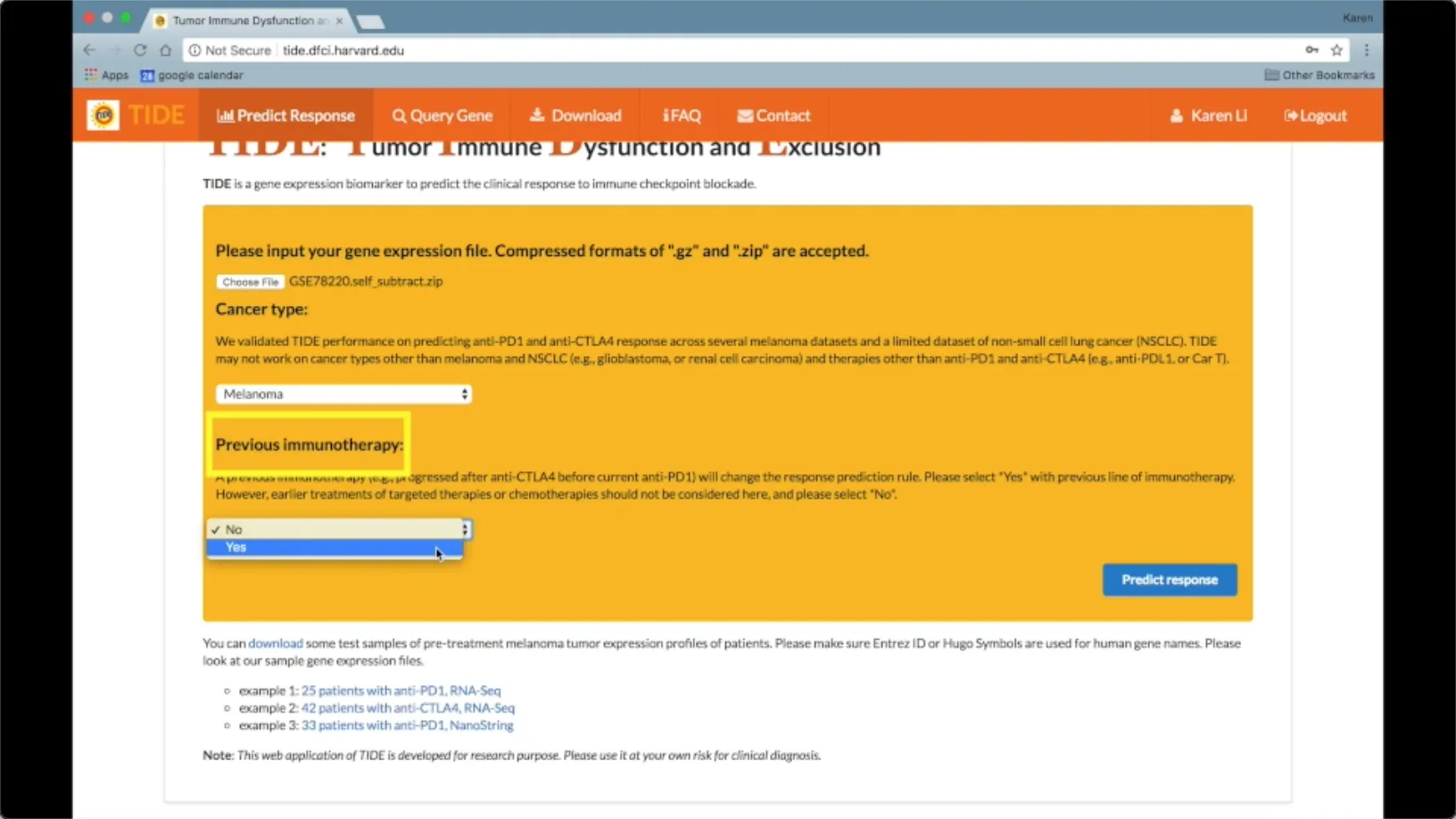
6、既往是否接受过免疫治疗
要选择是否既往接受过免疫治疗,因为接受了免疫治疗会改变免疫反应的情况。
结果解读
点击predict response之后就会得到结果,图片和表格文件均可导出至本地。
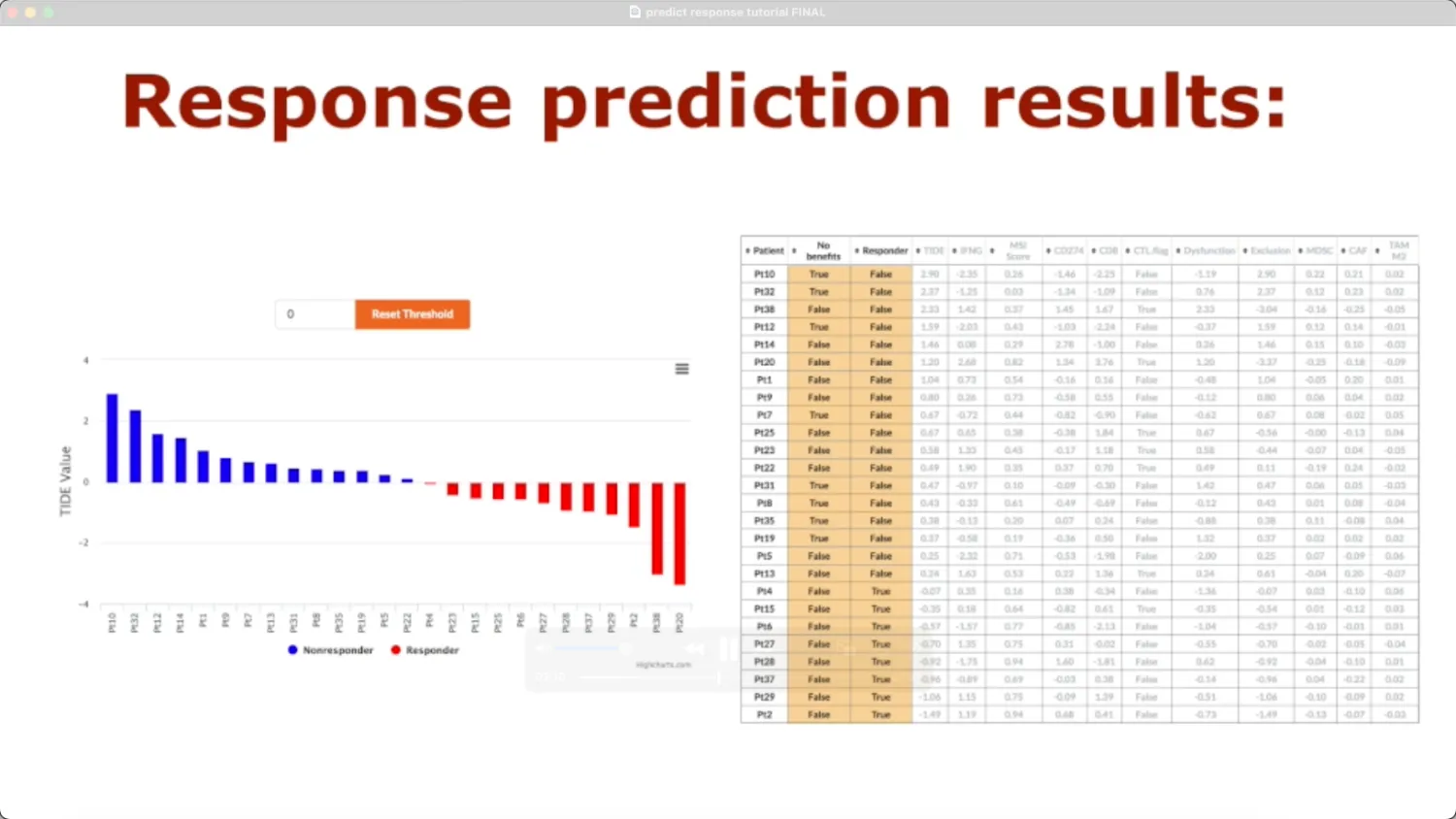
左边的柱状图:
中蓝色的代表Nonresponder,TIDE值为正(T细胞功能障碍和清除分值高),表示不能对免疫检查点抑制剂(ICB)产生反应。红色代表Responder,TIDE值为负(T细胞功能障碍和清除分值低),表示能够对ICB产生反应。
最上面的阈值可以自行调整(0-1),开发者建议研究者只关心预测Nonresponser的准确性的话,建议使用较高的阈值,比如1。如果研究者关注可能有反应的患者,建议使用较低阈值,比如0。
右边的表格:

No.benefit:预测是否对ICB存在反应,基于基因特征和干扰素γ信息,IFN-γ是有CTL分泌的哦。
Responder:预测是否对ICB存在反应,但仅基于基因特征。 TIDE:每个患者的预测分数,该列较高表明该患者具有较高的肿瘤免疫逃潜力,可能无法从ICB治疗中得到获益。
IFNG:6基因的IFN-γ标志物(IFNG,STAT1,IDO1, CXC10,CXCL9和 HLA-DRA)。
MSI score:微卫星不稳定,越高往往对ICB越敏感。
CD274和CD8(CD8A/B的平均表达量)就是这两个标志物的表达情况。
CTL.Flag: 表示T细胞和CTL的浸润水平,高浸润是True,低浸润是Flase。
Dysfuction:表示肿瘤患者的T细胞功能障碍情况。
Exclusion:表示肿瘤患者的T细胞清除/排除情况。
最后三列分别代表了MDSC/CAF/TAM-M2细胞(通常认为是免疫抑制相关的细胞) 与输入文件的pearson相关性。
分析步骤及流程
1、输入数据及处理
rm(list = ls())
load("~/Desktop/data.Rdata")
head(exp)[1:4,1:4]
# GSM1020099 GSM1020100 GSM1020101 GSM1020102
# RFC2 7.727199 5.109329 8.021387 7.880099
# HSPA6 7.601476 6.538490 6.340193 6.016021
# PAX8 5.910106 5.848390 6.124806 5.894079
# GUCA1A 3.577948 3.591067 3.566542 3.553448
# 数据的归一化处理,两种不同的代码
normalize <- t(apply(exp, 1, function(x)x-(mean(x))))
#normalize <- sweep(exp,1, apply(exp,1,mean,na.rm=T))
write.table(normalize, file = "exp.txt", sep = "\t",
row.names = T, col.names = TRUE, quote = FALSE)
2、得到结果
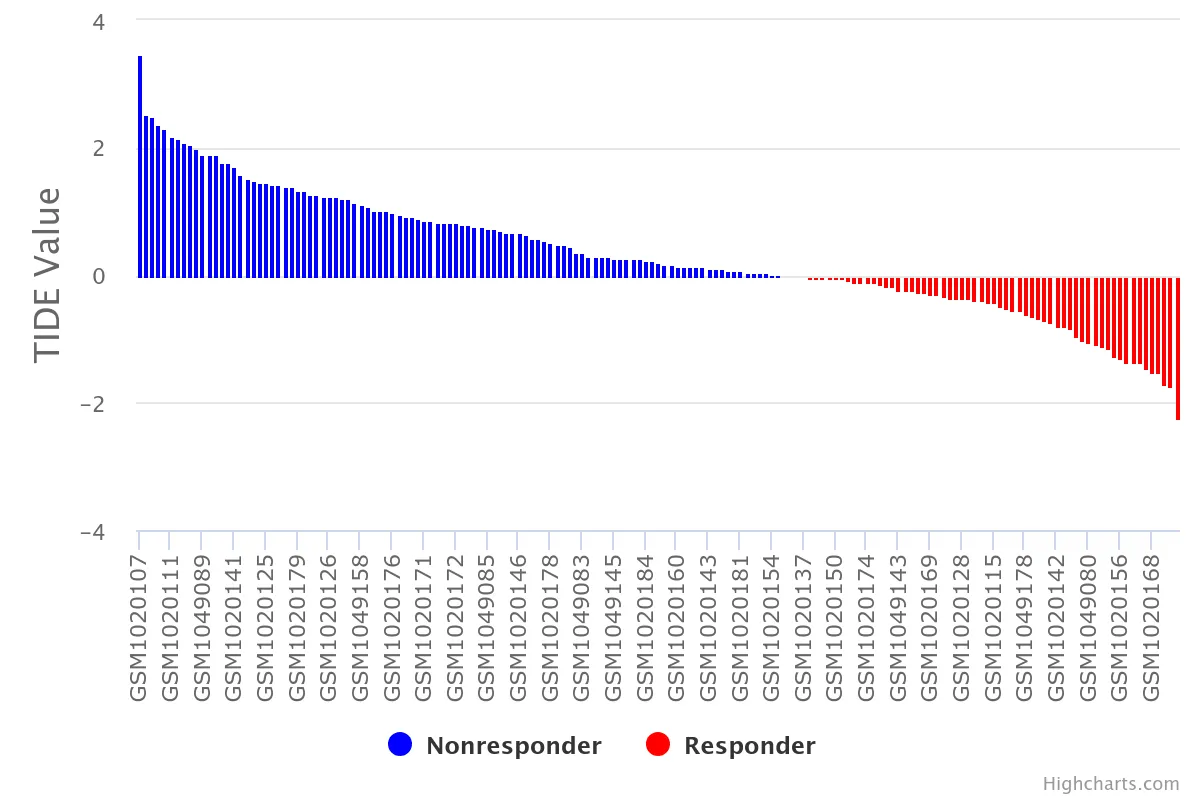

3、数据合并
# 把分组信息和TIDE信息合并
# meta是含有临床信息的表格,TIDE是从网站上获取的结果
meta <- read.csv("./meta.csv",row.names = 1)
TIDE <- read.csv("./exp_TIDE.csv",row.names = 1)
identical(rownames(meta),rownames(TIDE))
s <- intersect(rownames(meta),rownames(TIDE))
meta <- meta[s,]
TIDE <- TIDE[s,]
meta_TIDE <- cbind(meta,TIDE)
write.csv(meta_TIDE,"meta_TIDE.csv")
4、可视化处理
基础数据可视化
# 统计学检验一下
table(res$Responder,res$cluster)
f = fisher.test(table(res$cluster,res$Responder))
label = paste("fisher.test p value =",round(f$p.value,3))
label
# 网站中的柱状图
library(ggplot2)
library(dplyr)
res = arrange(res,desc(TIDE))
p1 = ggplot(res,aes(x = 1:nrow(res),
y = TIDE,
fill = Responder))+
geom_bar(stat = "identity") +
scale_fill_manual(values = c("#e04030","#6cb8d2"))+
xlab("patient")+
ylab("TIDE value")+
annotate("text", x = 40, y = -2, label = label,size = 10)+
theme_bw()+
theme(legend.position = "none") # 把P1的图注去掉了
########免疫反应与亚型
library(dplyr)
dat=count(res,cluster,Responder)
dat=dat%>%group_by(cluster)%>%
summarise(Responder=Responder,n=n/sum(n))
dat$Responder=factor(dat$Responder,levels=c("False","True"))
dat
library(ggplot2)
p2=ggplot(data=dat)+
geom_bar(aes(x=cluster,y=n,
fill=Responder),
stat="identity")+
scale_fill_manual(values=c("#e04030","#6cb8d2"))+
geom_label(aes(x=cluster,y=n,
label=scales::percent(n),
fill=Responder),
color="white",
size=4,label.size=0,
show.legend = FALSE,
position=position_fill(vjust=0.5))+
ylab("Percentage")+
theme_minimal()+
guides(fill = guide_legend(title = "Responder")) # 仅保留一个图例
library(patchwork)
p1+p2+plot_layout(widths=c(3,2),guides="collect")
ggsave('total.png',width = 20,height = 8)
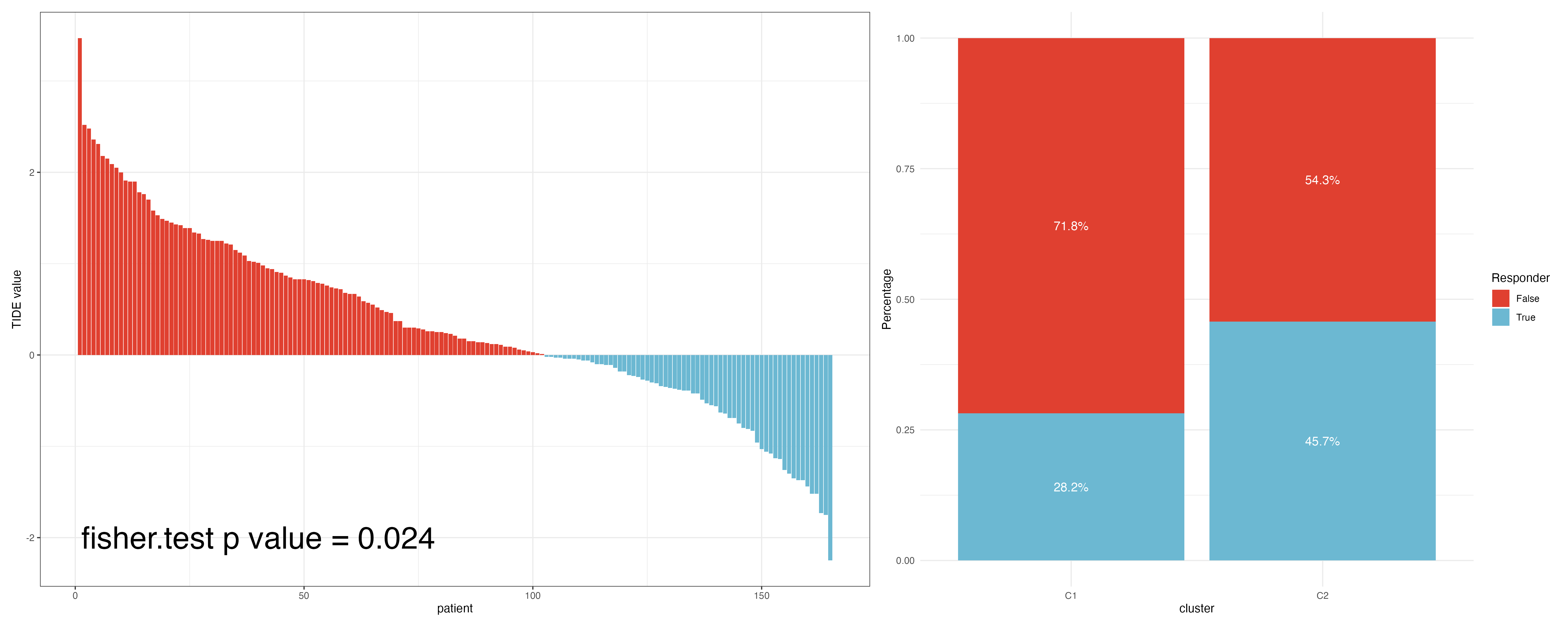
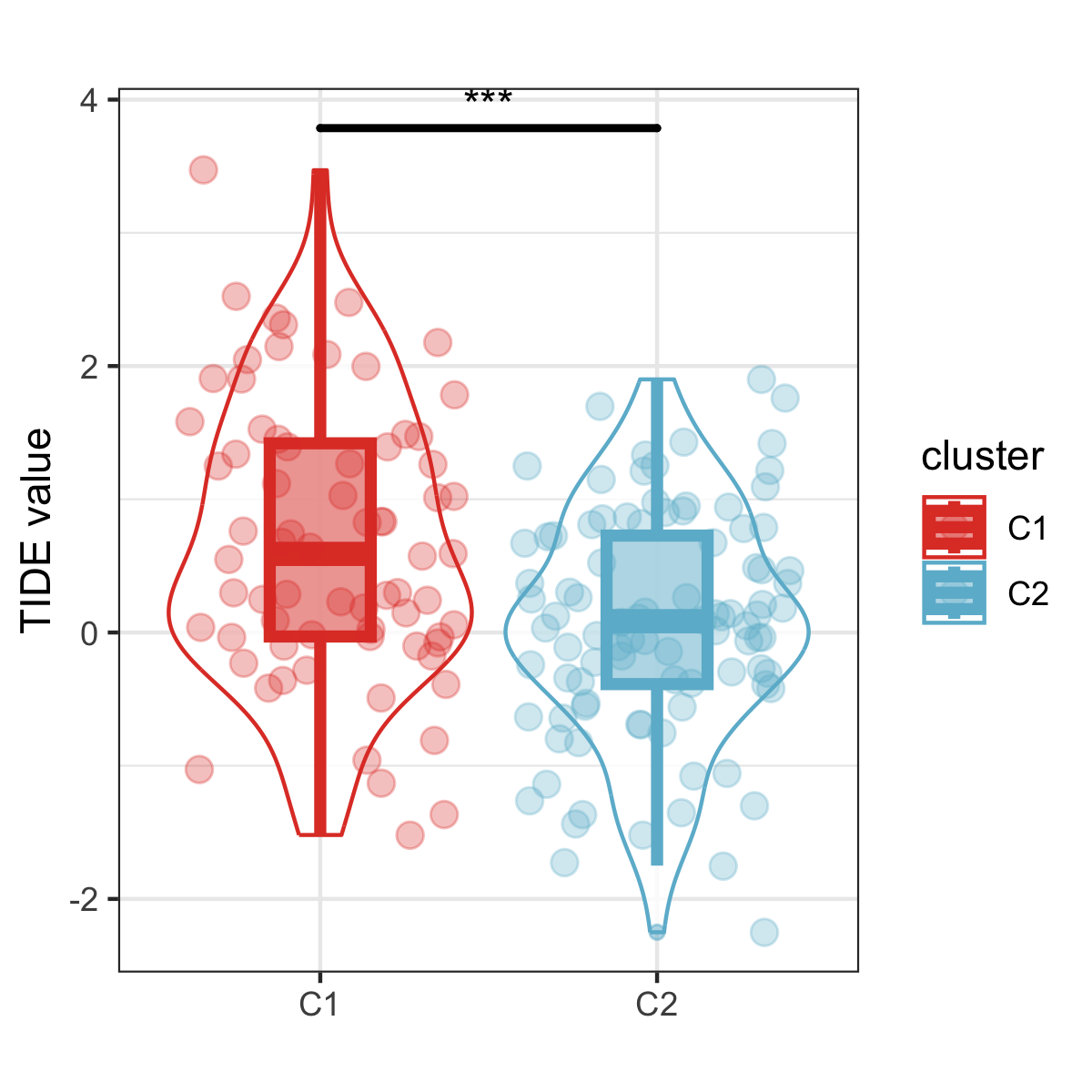
TIDE可视化
res$cluster <- factor(res$cluster,levels = c("C1","C2"))
png("TIDE_web.png",width = 1200,height = 1200,res = 300)
ggplot(data=res,aes(x=cluster,y=TIDE,colour = cluster))+ #fill参数不要设置,会不好看
geom_violin(#color = 'grey',
alpha = 0.8, #alpha = 0.8 参数控制着小提琴图的透明度。
scale = 'width',#小提琴宽度
#linewidth = 1, #外轮廓粗细
trim = TRUE)+ # trim = TRUE 参数控制着小提琴图的形状。
geom_boxplot(mapping=aes(x=cluster,y=TIDE,colour=cluster,fill=cluster), #箱线图
alpha = 0.5,
size=1.5,
width = 0.3)+
geom_jitter(mapping=aes(x=cluster,y=TIDE,colour=cluster), #散点
alpha = 0.3,size=3)+
scale_fill_manual(limits=c("C1","C2"),
values =c("#e04030","#6cb8d2"))+
scale_color_manual(limits=c("C1","C2"),
values=c("#e04030","#6cb8d2"))+ #颜色
geom_signif(mapping=aes(x=cluster,y=TIDE), # 不同组别的显著性
comparisons = list(c("C1","C2")), # 哪些组进行比较
map_signif_level=T, # T显示显著性,F显示p value
tip_length=c(0,0),#把向下的帽子去掉,分组数乘以2
y_position = c(3.5), # 设置显著性线的位置高度
size=1, # 修改线的粗细
textsize = 4, # 修改显著性标记的大小
test = "wilcox.test", # 检验的类型,可以更改
color = "black")+ # 设置显著性线的颜色
theme_bw()+ #设置白色背景
guides(fill = guide_legend(title = "cluster"), # 设置填充图例的标题
color = guide_legend(title = "cluster"))+ # 设置颜色图例的标题
labs(title = "", # 设置标题
x="",y= "TIDE value") # 添加标题,x轴,y轴标签
dev.off()
其他值可视化
library(tinyarray)
res$cluster <- factor(res$cluster,levels = c("C1","C2"))
colnames(res)
dat <- t(res[,c(12,14:16,18:22)])
head(dat)[1:4,1:4]
draw_boxplot(dat,res$cluster)+
facet_wrap(~rows,scales ="free") +
scale_fill_manual(values = c("C1" = "#e04030", "C2" = "#6cb8d2"))
ggsave("together.png",width = 12,height = 12)

参考资料:
1、TIDE:Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018 Oct;24(10):1550-1558.
2、生信菜鸟团:https://mp.weixin.qq.com/s/AHJCgY_341ZY5pmq6vSQCw
3、生信星球:https://mp.weixin.qq.com/s/-Vu6UcIesrC096aXRPRTyA
致谢:感谢曾老师、小洁老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -