
– https://doi.org/10.1038/s41576-023-00599-5
留意更多内容,欢迎关注微信公众号:组学之心


Single-cell genomics meets human genetics
单细胞基因组技术已经扩展到可以检测数千个个体的样本的程度。将大规模的单细胞信息与基因型数据相结合,能够把遗传变异与支撑人类生物学和疾病关键方面的细胞过程联系起来,助力对疾病诊断、风险预测和治疗解决方案的开发。但是,有效地整合大规模单细胞基因组数据、遗传变异和其他表型数据,前提是需要在数据生成和分析方法方面取得进步。
Introduction
背景:
全基因组关联研究 (GWAS)发现了大量与复杂疾病和人类特征相关的遗传变异,但大多数变异的生物学机制尚不明确,尤其是位于非编码区域的变异。表达数量性状基因座 (eQTL) 定位可以估计遗传变异(如SNP)与基因RNA水平的关联,将变异与调控的靶基因联系起来,有助于理解遗传变异的作用方式,识别可能参与疾病发病机制的基因,确定治疗干预的机会。
eQTL研究现状与未来:
目前,Genotype-Tissue Expression Consortium 在50多种人体组织中绘制了bulk eQTL景观,使用bulk转录组评估平均表达水平。但bulk eQTL研究在解决罕见细胞状态和动态状态方面存在局限,或者缺乏具有强效 FACS 抗体的表面蛋白。此外,一些瞬时或动态状态无法在体外重现。尽管组织水平 eQTL 在 GWAS 中富含与疾病相关的遗传变异,但只有 20-50% 的常见疾病等位基因与 eQTL 共定位,这表明许多变异通过细胞状态特异性机制影响生物学,而且有的eQTL效应可能仅在特定细胞类型或条件下检测到(如response eQTL),如果不采用全新的方法,就无法识别这些机制。
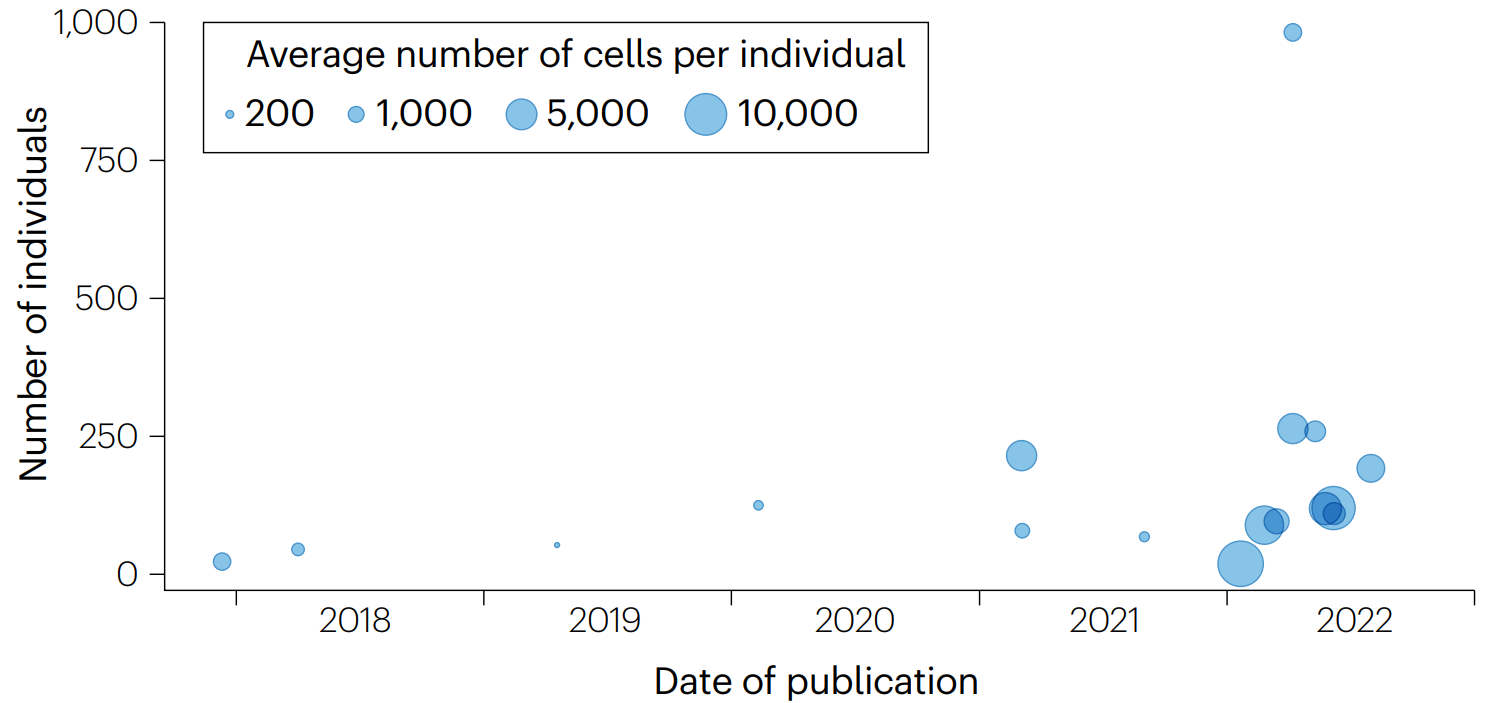
scRNA-seq技术结合遗传关联研究,推动了“单细胞遗传学”研究。sc-eQTL 研究解决了bulk数据无法解答的问题,如随细胞环境变化的eQTL和特定细胞状态下的基因调控。自2022年1月至12月,sc-eQTL研究发表数量增加了一倍以上。国际倡议(如单细胞 eQTLGen 联盟)在协调这一领域的研究努力。sc-eQTL研究提供了高分辨率表达图,有望为治疗开发提供有价值的信息。
综述内容:
- 回顾单细胞基因组学和人类遗传学及其交集。
- 回顾第一个sc-eQTL研究,展示bulk分析方法在单细胞数据中的应用可行性。
- 讨论与传统bulk RNA 测序相比的挑战。
- 介绍基于单细胞分辨率的新方法,如映射沿连续轨迹变化的eQTL。
- 展望该领域的未来方向,包括新的数据类型和集成策略,以及临床和治疗应用的转化。
1.关键领域的简要回顾
作者将单细胞遗传学定义为单细胞基因组学和人类遗传学交叉领域的新兴领域。这两个领域各有机遇、挑战和瓶颈。在这里,作者回顾了这一交叉领域的相关差距和协同作用,并介绍了为本综述提供必要背景的概念。
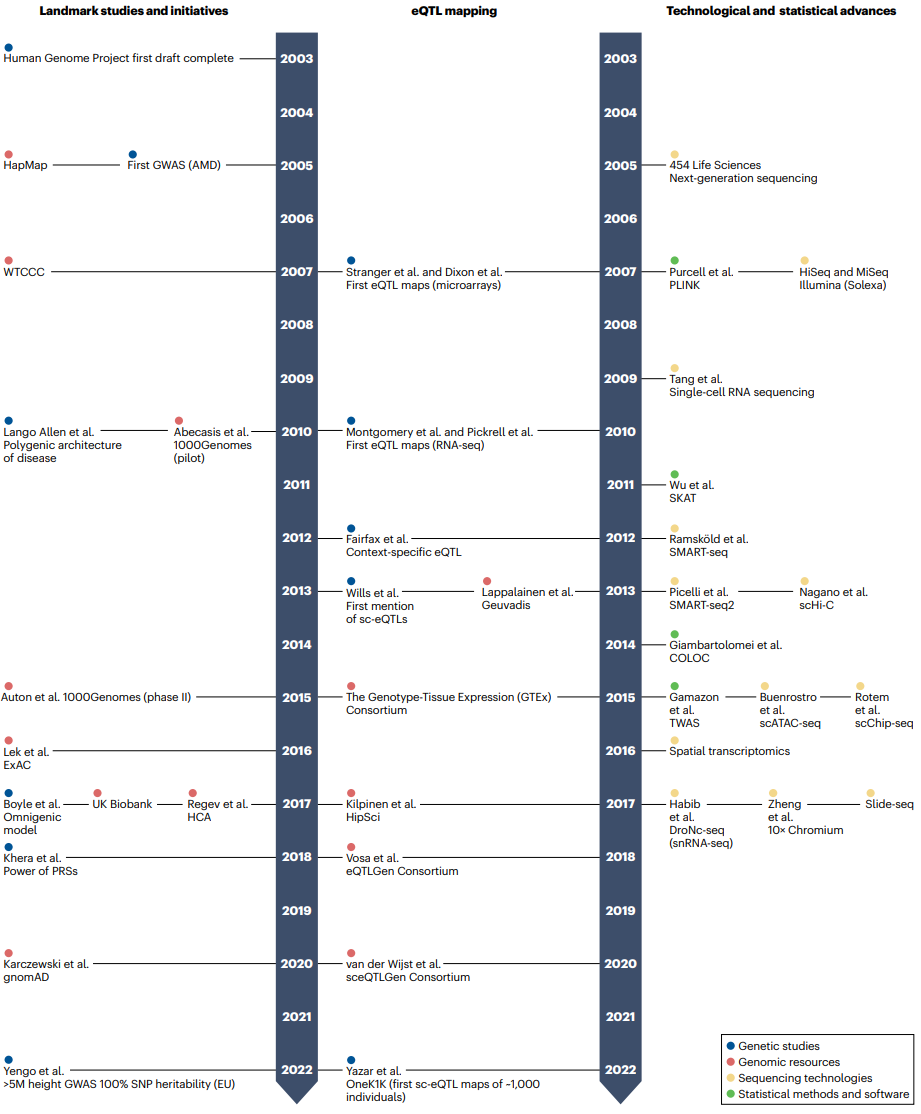
人类遗传学和单细胞基因组学的代表性研究成果(20 年的时间线):
1.1 单细胞基因组
scRNA-seq 技术的进展
单细胞 RNA 测序 (scRNA-seq) 是最常见的单细胞技术,自 2009 年以来迅速扩展,从最初的 8 个细胞到如今的 400 多万个细胞。基于液滴的技术是scRNA-seq目前最流行的方法,可以扩展到数万个细胞。单细胞被封装在微滴中,与独特条形码杂交的 mRNA 分子进行测序。基于板的技术(如 Smart-seq),将单细胞物理分离到 96 孔或 384 孔板中进行文库制备和全长转录本测序。此外,单核 RNA 测序是在分离单细胞有困难的情况下(如冷冻样本)的一种替代方法。

数据分析的新挑战和解决方案
单细胞数据分析产生了不同于bulk RNA 测序数据分析所面临的因素:例如,典型实验生成的大量配置文件、数据的稀疏性和一系列技术伪像。目前已经开发出新方法来应对这些挑战,此外还开发了特定的生物信息学工作流程(如 Cell Ranger)来处理原始数据,例如读取级质量控制、将读取分配给其细胞条形码和来源 RNA 分子(即“解复用”)、与参考基因组比对和量化。scRNA-seq 数据通常表示为分配给每个细胞中每个基因的测序读取(或分子,如果使用唯一分子标识符 (UMI))数量的整数矩阵。对于多个体汇集设计(特别适用于单细胞遗传研究),需要使用解复用方法来将细胞分配给来源个体(例如,demuxlet 和 vireo)。
预处理与标准化
预处理:包括检测和排除空液滴、双联体和环境 RNA;标准化调整细胞的总测序深度;对数转换和校正混杂因素(如技术批次和细胞周期效应)。
下游分析
- 降维:减少数据集的维数以降低计算负担和减少噪音,使用线性方法(如 PCA)和非线性变换(如 t-SNE 和 UMAP)。
- 细胞水平分析:确定细胞状态及其动态关系,包括聚类、细胞类型注释或轨迹推断。
- 基因水平分析:表征细胞状态的转录谱,包括差异表达或基因调控网络。
分析软件提供了全面的计算工具包,创建用户友好的单细胞工作流程和一致的数据对象。
常用工具包有 R 版本(如 Seurat 和 scran)和 Python 版本(如 Scanpy)。
1.2 遗传变异对分子表型的影响
基因组测序技术在遗传变异中研究的应用
测序技术的快速进步使得基因组测序项目的规模显著扩大,数十万个人类遗传变异得以表征。其中基因型芯片是低成本解决方案,能够系统地测量约 500,000 个“标记”位点的基因型,适用于大人群。而DNA 测序方法能够解决罕见和结构性遗传变异(群体次要等位基因频率 <1%),测序方法包括全外显子组测序和全基因组测序,使用短读或长读方法。
在严重的单基因疾病中,DNA 测序方法在研究和临床环境中的应用提高了基因诊断和疾病基因发现的速度。此外,对于复杂性状和常见疾病,GWAS 已识别出 400,000 多个遗传关联,并开发了多基因风险评分 (PRS),该评分结合了整个基因组的关联信号来预测个体的疾病风险。
功能基因组分析
遗传变异研究可以与功能基因组分析相结合,以直接评估个体变异的潜在生物学影响。最流行的方法是表达 (e)QTL 映射,类似的框架可用于 DNA 甲基化、蛋白质、组蛋白修饰、染色质可及性和剪接。
生物理论上预计大多数调控区域都靠近其目标,因此大多数 QTL 研究都集中在近端 (顺式) 映射上,例如,考虑基因内和周围的变异、甲基化位点或感兴趣的可及性峰值。相比之下,远端 (反式) QTL 映射考虑远端染色体间调控,但需要更大的样本量。
QTL 与 GWAS 的联系
QTL 研究的样本量远小于 GWAS。遗传对分子特征的影响通常大于对疾病风险的影响,现有样本量足以识别这些影响。此外,罕见变异研究较困难,主要局限于研究极端表型个体中的罕见变异,方法仍在发展中。
- 分子功能揭示:将 QTL 结果与 GWAS 结果联系起来,揭示与疾病相关的遗传变异的分子功能。
- 统计共定位:评估 QTL 是否与疾病位点一致。
- 孟德尔随机化:评估 QTL 对中间分子性状的影响是否是疾病的因果关系。
- 全转录组关联研究 (TWAS):利用 eQTL 信息推断 GWAS 病例和对照的基因表达,直接关联性状和基因。
2.使用pseudo-bulk counts 进行单细胞 eQTL 映射
测序成本的降低、完善的方法、处理流程、多路复用技术和批次效应消除方法使得单细胞基因组学(特别是转录组学)能够应用于大型基因型群体。此外,在单细胞遗传学研究中,使用来自同一个体的单细胞分子分析和基因型能够在细胞水平上评估遗传变异对分子表型的影响。在这里,文章专注于 sc-eQTL 研究,探讨在单细胞分辨率下测试遗传变异与基因表达变化之间的关联。
2.1 概念验证和早期细胞类型研究
早期研究与动机
2013年首次绘制sc-eQTL,研究动机源于发现对大量细胞的平均基因表达会掩盖一些基因表达表型(就是某些表型在细胞群中平均下来后观察不到,如转录爆发、噪声和动态表达波动)。虽然首次绘制的sc-eQTL仅限于15个淋巴母细胞系中的WNT通路基因,但研究证明了SNP与单个细胞间的转录变异和相关性相关,提供了概念的初步证明,强调了单细胞解析在遗传研究中的价值。2013年~2018年中,研究证明了转录组范围的sc-eQTL分析的可行性,利用单细胞在检测、解复用和聚类细胞方面的进展,专注于人类外周血中明确界定的免疫细胞类型。尽管样本量有限(<50个个体),这些研究发现了数十到数百个eQTL。
sc-eQTL 分析方法
初步方法:测量基因型队列中的单细胞基因表达,对表型相似的细胞进行聚类,将每个簇或细胞类型中每个基因的聚合表达与附近变异个体的基因型相关联。
pseudo-bulk eQTL分析:这种方法基于现有的bulk eQTL流程,使其在计算上可扩展到逐渐增大的队列(目前最大的sc-eQTL研究考虑了近1,000个个体)。该方法与更复杂的组织单细胞表型的方法结合(例如拟时序分析,即沿轨迹的bins或高分辨率细胞状态簇),允许该方法扩展到更多异质组织和颗粒细胞类型。
2.2 最初为 bulk eQTL mapping 设计的方法
传统关联方法的局限性
最初的 sc-eQTL 研究使用了设计用于bulk eQTL 映射和基因型与连续性状关联测试的传统方法。这些方法假设:表型在所有样本中的分布近似于高斯分布;每个个体只有一个表型观察值。但是单细胞表达数据稀疏且包含多个观察值,这两个假设不适用。
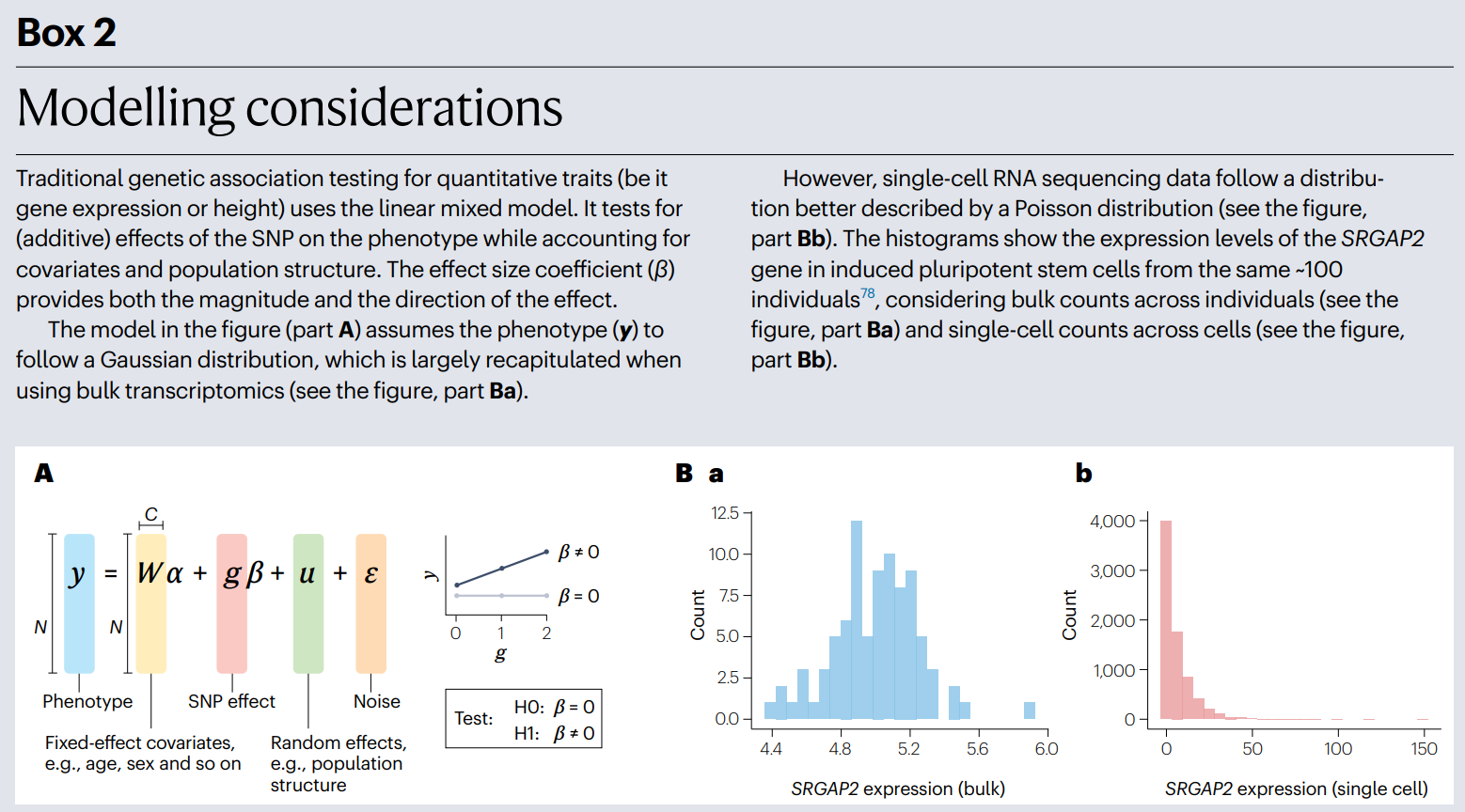
pseudo-bulk 策略
为克服上述局限,许多研究使用 pseudo-bulk 策略,将多个细胞的基因表达水平聚合,模拟单个 bulk 样本。pseudo-bulk 样本中基因的表达通常是基因raw count的总和或感兴趣的细胞类型中个体细胞中基因标准化表达的平均值(使用 scRNA-seq 数据更精确地定义)。
单细胞 eQTL 研究的挑战
如bulk研究一样,某些非遗传因素(协变量)可能会干扰或掩盖等位基因对基因表达的真实影响,与等位基因效应混淆。需要采用方法来检测和校正这些协变量,以确保等位基因效应的准确性和可靠性。因此,迁移了bulk分析中用于校正协变量的方法,如主成分分析和表达残差的概率估计 (PEER) 到pseudo-bulk分析。
此外,单细胞研究面临其他挑战,例如每个个体的细胞计数变化(与pseudo-bulk 计数的置信度成反比)或多实验研究设计的批次效应,这可能会导致实验池之间基因表达的系统差异。sc-eQTL 模型可以通过考虑这些实验因素以及额外的固定或随机效应来提高功效。对于伪批量 sc-eQTL 研究,有许多可能的单细胞计数标准化和聚合以及协变量校正策略
探索使用单细胞数据的额外信息
这些数据可以测量遗传变异与细胞间基因表达变异之间的关联。

变异性增加可能反映出表达稳定性的缺乏和进入极端致病状态的倾向增加,或者可能揭示基因-环境 (GxE) 与未测量的环境和背景的相互作用。尽管少数研究提出了从单细胞数据映射此类“variance eQTL”的方法,但由于样本量不足以及基因表达的平均值和方差之间的混杂相关性,研究结果较差。这些早期利用单细胞分辨率数据进行遗传关联模型的尝试为使用单细胞分辨率数据建模 eQTL 的新视角奠定了基础。随着单细胞遗传研究规模的扩大,以及更复杂的方法的出现,单细胞variance eQTL 研究将变得更加容易处理。
3.单细胞分辨率 eQTL 建模
早期的 eQTL 研究将同一细胞类型的细胞离散化和聚合,以方便进行统计建模和解释。然而,高分辨率单细胞数据通常会揭示离散群体内的异质性,这促使人们以单细胞分辨率对 eQTL 进行建模。在这里,文章描述了第二代 sc-eQTL 模型,该模型采用连续框架来利用细粒度的单细胞分辨率数据。
3.1 单细胞模型改进了细胞状态依赖的 eQTL 映射
连续表型与中间状态
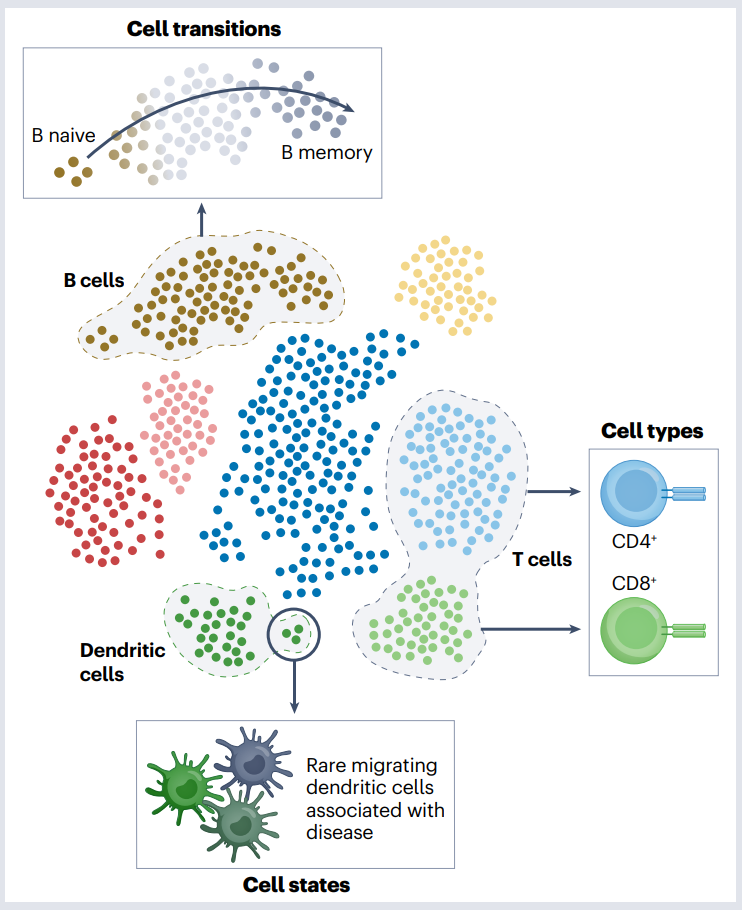
高分辨率分子测量(如转录组学)被用于定义和表征单细胞表型,揭示了细胞亚群类型、连续表型变化及中间状态。
连续表型例子,强调了对细胞状态进行更细致和连续定义的必要性:
- 人类 T 细胞的 scRNA-seq 研究发现跨多个 T 细胞亚谱系的细胞毒性连续体。
- 发育过程中,确定中胚层或内胚层命运之前的中内胚层状态。
- 类风湿性关节炎中的 NOTCH3 信号传导导致成纤维细胞向炎症状态转变。
模拟遗传调控动态变化
使用单细胞分辨率数据定义连续状态后,可以模拟遗传调控如何沿着这些轨迹动态变化。
这些模型不是将个体视为观察结果,而是将每个细胞视为其自身对基因表达的观察结果。
混合效应相互作用模型:其中包括随机效应,以解释来自同一个体的细胞的非独立性(如果不加以考虑,可能会增加假阳性率),以及细胞状态和基因型之间的相互作用项,以模拟基因型对表达的状态依赖性影响。这些第二代模型映射“dynamic” eQTL,评估不同基因型等位基因对沿连续轴动态变化的性状的影响。
生成统计(“topic”)模型:识别基因型和 scRNA-seq 谱之间的共同变异,识别离散细胞类型中的顺式 eQTL 和反式 eQTL。
分解方法:识别离散细胞类型中共同或特定的遗传效应。
3.2 单细胞表达数据的稀疏性和非正态性
由于采样不完整以及细胞内转录本存在真正的生物学差异,单细胞数据是稀疏的(包含许多 0)。因此,单细胞测量值不能很好地用线性回归模型假设的高斯分布来描述。在bulk 转录组中一起测定的大量细胞(以及在一定程度上 pseudo-bulk 聚合测量值)意味着标准化表达谱可以近似为高斯分布,但这并不适用于单细胞谱。
离散计数分布可以更好地描述这些数据。尽管单细胞谱很稀疏,但已证明不会出现零膨胀。泊松分布提供了一种可解释的单细胞计数模型,该模型已在最近的研究中使用,包括 泊松混合效应回归,泊松降秩回归模型。在某些情况下,根据基因表达分布,更多参数化的负二项式或多项式模型可能是合适的替代方案;P 值膨胀的零检验可以指导这些选择。
3.3 单独建模每个细胞与可扩展性
单独建模 vs pseudo-bulk测量
单独建模每个细胞:需要数十万个细胞的数据集,而不是像对相同数量个体的pseudo-bulk研究那样需要数百个样本。一种方法是将少于 10 个表型相似的细胞分组为“meta细胞”,这种聚合比将数千个细胞分组为一个簇或将数百个细胞分组为拟时序bins的破坏性更小,仍然适用于 eQTL 建模。
可扩展性与基础设施
随着未来研究中唯一个体数量的增加,方法必须具备可扩展性,并与高速计算和数据存储基础设施兼容。
学习与借鉴:
- TensorQTL:用于 QTL 映射的 Matrix eQTL 的图形处理单元实现。
- Hail:基于云的可扩展遗传学工具实现。
- 图形处理单元 (GPU):一些计算资源密集的 sc-eQTL 方法,如 scTBLDA,已经开始利用 GPU。
- 方法也可能受益于跨计算资源、基于云的系统以及代数和数值近似的并行化。
4.新机遇
当前的 sc-eQTL 映射范例只能提供有限的窗口来了解遗传学和细胞功能的整体情况。我们需要新的技术进步、更大规模的研究以及相应的分析和计算方法来扩展我们的视野。特别是,我们设想研究探索更多的分子特征(超越基因表达)、更多类型的遗传变异(超越常见的 SNP)以及更多关于个体的信息(例如人口统计、疾病史和环境暴露)。此外,我们希望从越来越多样化的群体中收集数据,包括来自不同祖先的个体、患有疾病的个体和许多不同(与疾病相关的)人体组织的数据。随着这些丰富的数据变得可用,需要新的分析和计算方法来整合跨数据模式(例如染色质可及性、表达和蛋白质水平)和分辨率(从细胞到组织再到个体)的信息,模拟特定于上下文的动态效应并预测与人类生物学和健康相关的结果。
4.1 新的数据形式
整合多种方法
对于可以测量的任何分子表型,都可以整合基因型信息来映射与表型相关的 QTL。我们能够更有效地测量和计算分析单细胞分辨率下的更多分子特征,因此可以在单细胞分辨率下探究更多细胞状态和分子过程的遗传学(例如,单细胞染色质可及性 QTL)。
多组学技术使我们能够在同一个细胞内分析多种数据模式:
- scNMT-seq:测量染色质可及性、DNA 甲基化和基因表达。
- CITE-seq:测量基因表达和表面蛋白水平。
整合多种模式可提供对同一细胞表型的多种视角,从而能够更高分辨率地定义细胞状态以模拟dynamic sc-eQTL,并提供多种表型来模拟与遗传变异的关系。这种整合任务被描述为“垂直整合”,细胞是跨模式的共同纽带。
样本量增加与新技术
适合分析的变异类别:包括罕见变异、重复、插入和缺失以及结构变异。
疾病相关性:这些变异与疾病有关,但在全组织水平上的分子表型影响分析有限。
单细胞水平的全面分析:使用单细胞模型和精确定义的细胞状态进行系统关联研究,全面捕捉这些变异在分子水平上的影响。
4.2 多样化群体
结合遗传学与分子测量
为了了解生物学背后的机制,需要将遗传学与不同自然扰动下的分子测量和细胞状态联系起来。可以通过分析数千名个体的细胞,这些个体具有已知的基因型和部分特征的“环境”信息,包括:
- 生活方式(如吸烟状况、饮食和污染)
- 人口统计学特征(如性别、年龄、地理和种族)
- 生物医学特征(如医疗和疫苗接种史、疾病状态和进展、药物)
环境和协变量的影响
- 纳入协变量:将这些不同变异源纳入单细胞遗传学研究,更好地理解遗传学与细胞水平变化的相互作用。
- 已证实的关系:协变量与细胞状态组成之间的关系已被证实,将其纳入 sc-eQTL 模型为动态 eQTL 提供丰富背景。
- 大规模数据集:允许研究 GxE 相互作用及其对分子特征的影响。
祖先多样性的考虑
研究需要考虑祖先多样性,以便研究不同人群中的致病等位基因、调控变异模式、细胞状态和活性途径。当前大多数研究的局限在欧洲人群,未能直接转化为非欧洲个体的研究成果。不同人群可能具有不同的疾病等位基因和变异模式,研究这些人群有助于更精确地定位动态 eQTL。目前已有一些祖先多样化的研究:
- 非欧洲人群研究:一些研究已在秘鲁、约鲁巴和非洲裔美国人群中进行。
- 大规模队列:正在生成来自亚洲、非洲和拉丁美洲的大规模多样性人群数据集,如:亚洲免疫多样性图谱、非洲血统免疫细胞图谱、拉丁美洲多样性人类细胞图谱
发展鲁棒的遗传算法
为了充分利用这些数据,需开发对多血统数据具有鲁棒性的遗传算法,用于关联测试、精细映射和荟萃分析。
4.3 研究疾病组织背景
组织特异性疾病与基因调控
许多疾病具有组织特异性表现,因此在疾病组织环境中研究遗传变异对基因调控的影响至关重要。但目前大多数 sc-eQTL 研究局限于易获取的组织(如皮肤和血液)或细胞系(如 iPS 细胞),少数研究涉及其他组织(如大脑),限制了我们了解疾病相关组织中基因调控的能力。
- 神经退行性疾病:如帕金森病,缺乏来自特定脑细胞(如多巴胺能神经元)的数据,导致研究困难。
- 环境特异性调控:即使在血液中可以找到的细胞类型,在组织中的环境也不同,可能受到不同的遗传调控。
- 组织处理难度:组织需要处理、冷冻和分解,增加了研究难度。
未来展望
大规模单细胞数据生成项目:如人类细胞图谱,可能解决关键问题,提供更全面的数据。
疾病相关基因调控研究的新途径。
患病个体的数据收集:从患有疾病和其他感兴趣特征的基因型个体获取单细胞分析数据。例如,Perez 等人在系统性红斑狼疮患者的各种血细胞类型中绘制了 sc-eQTL。
干细胞和类器官模型:通过将干细胞分化为感兴趣的细胞类型并培养类器官模型,解决难以获取的组织和疾病表型个体的基因型单细胞群的不足。
“细胞村”概念:扩大干细胞研究范围,探索与疾病相关的细胞类型和基因型中的基因调控。
空间 eQTL
空间转录组学技术以近细胞分辨率记录细胞的原位位置及其 RNA 表达谱,技术迅速改进,分辨率更高、成本更低、保真度更高、实施更易。空间 eQTL 研究能够了解基因调控如何与组织结构相互作用导致疾病。
新混合建模策略:将传统分析(如差异表达)扩展到空间转录组学,可能对 eQTL 模型有用。随着计算工具和空间技术的发展,绘制跨空间坐标变化的 eQTL 将成为可能。
5.助力疾病的理解与临床效益
eQTL 可以洞察与疾病相关的遗传变异的作用模式,包括它们所调控的基因、作用的方向以及它们起作用的细胞状态,这对于理解疾病过程以及最终帮助药物开发具有重要意义。
5.1 单细胞遗传学用于识别与疾病相关的细胞类型
识别组织与细胞环境新模式
开发细胞状态特异性 sc-eQTL 模型,结合现有疾病等位基因知识与细胞环境中的调控靶标,推断疾病等位基因可能最具破坏性的环境,了解与疾病表型最相关的组织、细胞类型和细胞状态,增进临床理解。开发针对受许多遗传变异影响的复杂性状的方法,整合大量组织特异性 eQTL 效应,优先关注疾病最相关的组织区域。
实例:Kundu 等人通过 eQTL 映射精细定位与疾病相关的致病变异,发现炎症性肠病的 ITGA4 基因座在单核细胞中活跃。
解析亚细胞类型
单细胞解析的 eQTL 图谱提供亚细胞类型解析。
实例研究:结合帕金森病和精神分裂症 GWAS 结果,使用 iPS 细胞衍生的多巴胺能神经元中的 sc-eQTL,确认和识别相关基因。
单细胞数据的其他方法
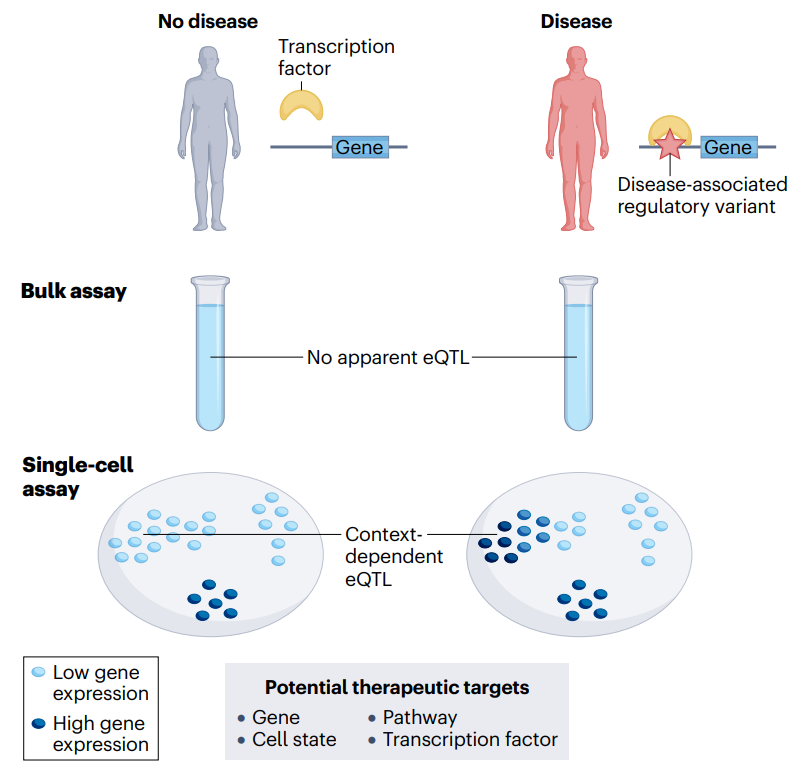
变异基因表达关联:使用不同策略将疾病相关变异与基因联系,估计与疾病相关的更精确的细胞类型。
单细胞疾病相关性评分 (scDRS):估计单个细胞与多基因疾病风险的关联,将 PRS 框架转化为单细胞水平。
单细胞分子 QTL 结果可能有助于构建类似的预测因子,利用单细胞分子 QTL 结果,可以进一步考虑遗传变异对细胞类型特异性的调控作用。
CONTENT 方法:使用上下文特异性 eQTL 来识别与疾病相关的基因,量化疾病遗传性的上下文特异性部分。
5.2 在临床中的潜能
临床应用的潜力与挑战
药物靶点与临床有效性:具有遗传证据的药物靶点在临床上有效的可能性是其他药物的两倍。但目前sc-eQTL 结果尚未能直接转化为临床应用,需要克服许多关键步骤。
疾病异质性与患者分层
- 异质性反映:复杂疾病的异质性可能反映潜在的遗传和机制差异。
- 分层工具:遗传(PRS)和基于表达的方法(体细胞和单细胞)用于根据疾病风险分层患者,并划分为疾病亚型。
- 贝叶斯混合建模:一种用于对组织特异性体细胞 eQTL 变体的性状关联进行优先排序的方法,识别与大脑、脂肪组织或肌肉中的基因调控最相关的患者亚群。
- sc-eQTL 的潜力:使用 sc-eQTL 研究可能在细胞类型和亚细胞类型分辨率下实现类似的患者分层,有助于根据疾病预后和最佳治疗策略划分亚组。
功能注释与诊断率提升
- 功能注释不完整的限制:DNA 测序对单基因疾病患者的诊断效用有限。
- 功能基因组分析的优势:临床组织样本的功能基因组分析提高了诊断率,特别是通过bulk RNA 测序识别致病的基因表达或剪接变化。
- 单细胞方法的潜力:对涉及特定疾病相关细胞状态生物学的基因组区域提供更准确的功能预测;识别影响转录结构或在可及组织中罕见的细胞类型中表达的变异,提高诊断率。





![洛谷 P1035 [NOIP2002 普及组] 级数求和 题解](https://i-blog.csdnimg.cn/direct/638c6c6eaf304409ba8b36c49f643e1b.png)














