#安装软件包
> if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
> BiocManager::install("limma")
> BiocManager::install("org.Hs.eg.db")
> BiocManager::install("DOSE")
> BiocManager::install("clusterProfiler")
> BiocManager::install("enrichplot")
#加载软件包
> library(limma)
> library(org.Hs.eg.db)
> library(clusterProfiler)
> library(enrichplot)
#设置变量
> gene="CRTAC1"
> expFile="combined_RNAseq_counts.txt"
> gmtFile="c5.go.v7.4.symbols.gmt"
> rt=read.table(expFile, header=T, sep="\t", check.names=F)
> head(rt)
head(rt)
id TCGA-E2-A1L7-11A TCGA-E2-A1L7-01A TCGA-AR-A0U0-01A TCGA-BH-A28O-01A TCGA-A2-A0D4-01A TCGA-E9-A1R4-01A TCGA-AO-A1KQ-01A
TCGA-AC-A62V-01A TCGA-D8-A143-01A TCGA-A2-A0SV-01A TCGA-AN-A0XW-01A TCGA-D8-A1XV-01A TCGA-A2-A4RW-01A TCGA-A7-A0CD-01A TCGA-E2-A1IG-11A
TCGA-D8-A1XB-01A TCGA-C8-A134-01A TCGA-BH-A0BS-11A TCGA-AR-A2LE-01A TCGA-A2-A0CO-01A TCGA-E9-A1NA-11A TCGA-AN-A0AK-01A TCGA-E9-A1NA-01A
TCGA-A7-A0DA-01A TCGA-E2-A572-01A TCGA-A2-A259-01A TCGA-BH-A28Q-01A TCGA-E2-A1IO-01A TCGA-AQ-A7U7-01A TCGA-AN-A0FD-01A TCGA-A8-A07G-01A
TCGA-AO-A0JL-01A TCGA-B6-A0IM-01A TCGA-B6-A0IP-01A TCGA-GM-A2DF-01A TCGA-A2-A25B-01A TCGA-BH-A0B0-01A TCGA-AO-A0JD-01A TCGA-AN-A0FL-01A
TCGA-E2-A14V-01A TCGA-AN-A0FF-01A TCGA-C8-A138-01A TCGA-E2-A14R-01A TCGA-AC-A2BM-01A TCGA-A1-A0SP-01A TCGA-A2-A0CQ-01A TCGA-A8-A08J-01A
TCGA-BH-A6R8-01A TCGA-E9-A1QZ-01A TCGA-A8-A0AB-01A TCGA-BH-A0H9-11A TCGA-AC-A3W7-01A TCGA-B6-A0IE-01A TCGA-A8-A07I-01A TCGA-BH-A0BQ-11A
> rt=as.matrix(rt)
> rownames(rt)=rt[,1]
> exp=rt[,2:ncol(rt)]
> dimnames=list(rownames(exp),colnames(exp))
> data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
> data=avereps(data)
> data=data[rowMeans(data)>0,]
> group=sapply(strsplit(colnames(data),"\\-"), "[", 4)
> group=sapply(strsplit(group,""), "[", 1)
> group=gsub("2", "1", group)
> data=data[,group==0]
> data=t(data)
> rownames(data)=gsub("(.*?)\\-(.*?)\\-(.*?)\\-.*", "\\1\\-\\2\\-\\3", rownames(data))
> data=t(avereps(data))
> dataL=data[,data[gene,]<=median(data[gene,]),drop=F]
> dataH=data[,data[gene,]>median(data[gene,]),drop=F]
> meanL=rowMeans(dataL)
> meanH=rowMeans(dataH)
> meanL[meanL<0.00001]=0.00001
> meanH[meanH<0.00001]=0.00001
> logFC=log2(meanH)-log2(meanL)
#排序
> logFC=sort(logFC,decreasing=T)
> genes=names(logFC)
> gmt=read.gmt(gmtFile)
#GESA分析
> kk=GSEA(logFC, TERM2GENE=gmt, pvalueCutoff = 1)
> kkTab=as.data.frame(kk)
> kkTab=kkTab[kkTab$pvalue<0.05,]
> write.table(kkTab,file="GSEA.result-GO.txt",sep="\t",quote=F,row.names = F)
> termNum=5
> if(nrow(kkTab)>=termNum){
showTerm=row.names(kkTab)[1:termNum]
gseaplot=gseaplot2(kk, showTerm, base_size=8, title="")
pdf(file="GSEA-GO.pdf", width=10, height=8)
print(gseaplot)
dev.off()
}

> my=read.table("my.txt", header=T, sep="\t", check.names=F)
> my=as.matrix(my)
> rownames(my)=my[,1]
> mys=my[,2:ncol(my)]
> showmy=row.names(mys)
> myplot=gseaplot2(kk, showmy, base_size=8, title="")
> pdf(file="GSEA-GO-myself.pdf", width=10, height=8)
> print(myplot)
> dev.off()

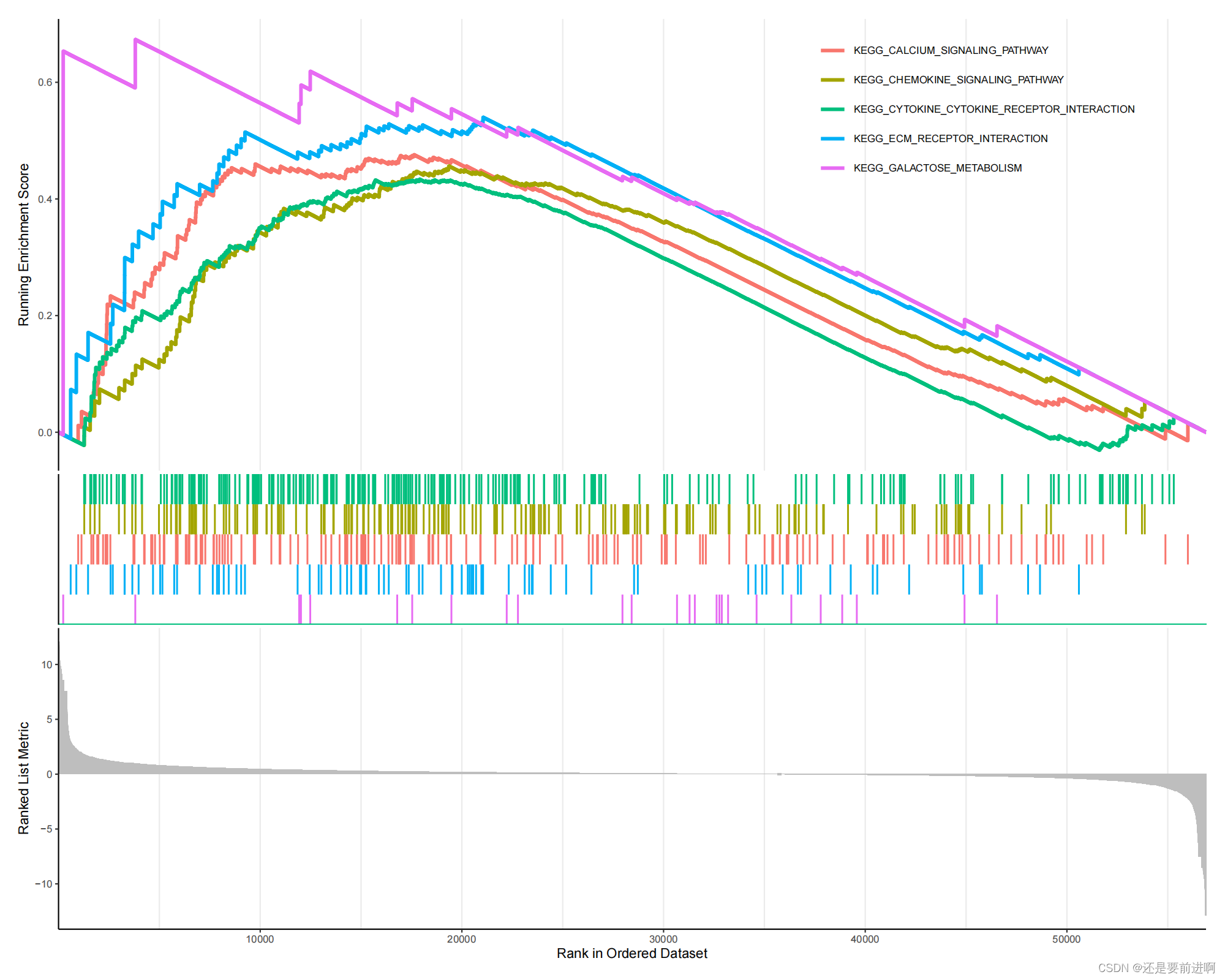
> gmtFile="c2.cp.kegg.v7.4.symbols.gmt"
> gmt=read.gmt(gmtFile)
> kk=GSEA(logFC, TERM2GENE=gmt, pvalueCutoff = 1)
> kkTab=as.data.frame(kk)
> kkTab=kkTab[kkTab$pvalue<0.05,]
> write.table(kkTab,file="GSEA.result-KEGG.txt",sep="\t",quote=F,row.names = F)
> termNum=5
> if(nrow(kkTab)>=termNum){
showTerm=row.names(kkTab)[1:termNum]
gseaplot=gseaplot2(kk, showTerm, base_size=8, title="")
pdf(file="GSEA-KEGG.pdf", width=10, height=8)
print(gseaplot)
dev.off()
}
一起学习交流。