长链非编码RNA(Long noncoding RNA)是一类长度大于200 nt,不编码蛋白质的RNA。LncRNA的功能多样,如图1所示,A:充当蛋白质和染色质的连接因子,引起染色质重构(chromatin remodeling);B:充当miRNA海绵;C:充当“scaffold”脚手架;D:与转录因子结合,引起转录激活;E:把转录因子拉走,引起转录抑制;FGH:与mRNA结合抑制翻译、调节剪切,及降解mRNA等。
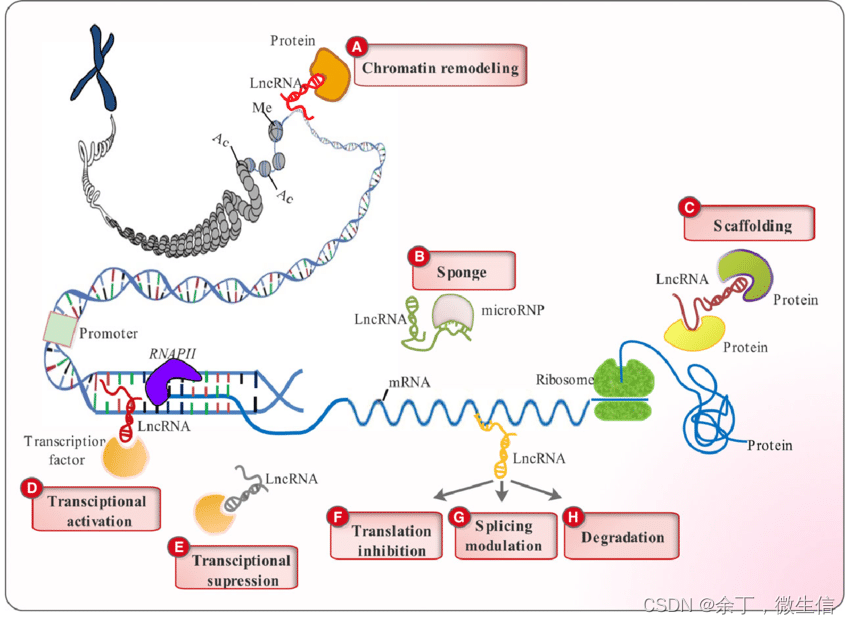
图1. lncRNA功能
由于lncRNA发现较晚,功能研究不完善,因此,我们可以以mRNA为桥梁对lncRNA的功能进行推断和研究。通常将lncRNA的功能分成cis和trans两种(图2)。Cis作用的原理是lncRNA的功能与其临近mRNA相关,可以以lncRNA基因组坐标上下游100 kb的mRNA作为lncRNA的靶基因进行研究。而trans作用的原理是lncRNA与其共表达的mRNA相关,可以根据表达量计算相关性以预测lncRNA的靶基因(一般要求样品数超过6个),从而研究lncRNA的功能。

图2. Cis vs trans
相关系数(correlation coefficient)
相关系数用来衡量两个变量X和Y间的相关性。相关性包括:线性相关和非线性相关。
Pearson相关系数用来衡量两个变量X和Y之间的线性相关关系。常用r表示,取值范围为[-1,1]。其中负的表示负相关,正的表示正相关。值越大相关性越强。
而spearman秩相关系数用来衡量两个变量间的非线性相关关系。是一个非参数度量。常用rho(ρ)来表示。取值范围也是[-1,1]。
常见的相关性标准为:不相关:0-0.1 ;低相关:0.1-0.3;中等相关:0.3-0.5;显著相关0.5-1.0。应参考具体使用场景进行判断。
虽然pearson相关系数最常用,然而它受数据分布的影响,对异常值敏感,需要数据服从近似正态分布才能使用。然而,我们遇到的数据是非常复杂的,往往并不符合线性相关,因此,越来越多的研究者使用spearman秩相关系数(Spearman’s rank correlation coefficient)计算两个变量间的相关性。注意:相关性不隐含因果关系。
在lncRNA-mRNA共表达相关系数计算中,pearson相关系数和spearman相关系数都有使用。例如在《Genome-wide analysis of lncRNAs, miRNAs, and mRNAs forming a prognostic scoring system in esophageal squamous cell carcinoma》文章中,作者写道“The correlation between prognostic lncRNA and mRNA expression profiles was analyzed by Spearman method, and the lncRNA-mRNAs pairs that the absolute value of correlation coefficients > =0.4 and p < 0.05 were selected to construct the co-expression network”,使用的是Spearman相关系数,rho阈值0.4,pvalue阈值0.05。而在《Genome-wide analysis of differentially expressed lncRNAs and mRNAs in primary gonadotrophin adenomas by RNA-seq》文章中,作者写道“The network is based on Pearson correlation coefficient (the absolute value of PCC ≥ 0.80, p-value < 0.001)”,使用的是pearson相关系数,r阈值0.8,pvalue阈值0.001。
今天,我们来计算相关系数,并使用cytoscape软件绘制网络图。
1.打开相关系数计算页面
首先,使用浏览器(推荐chrome或者edge)打开lncRNA-mRNA pearson、spearman相关系数计算页面。左侧为常见作图导航,中间为数据输入框和可选参数,右侧为描述和结果示例。
http://www.bioinformatics.com.cn/basic_lncrna_mrna_pearson_spearman_coexpression_analysis_t013
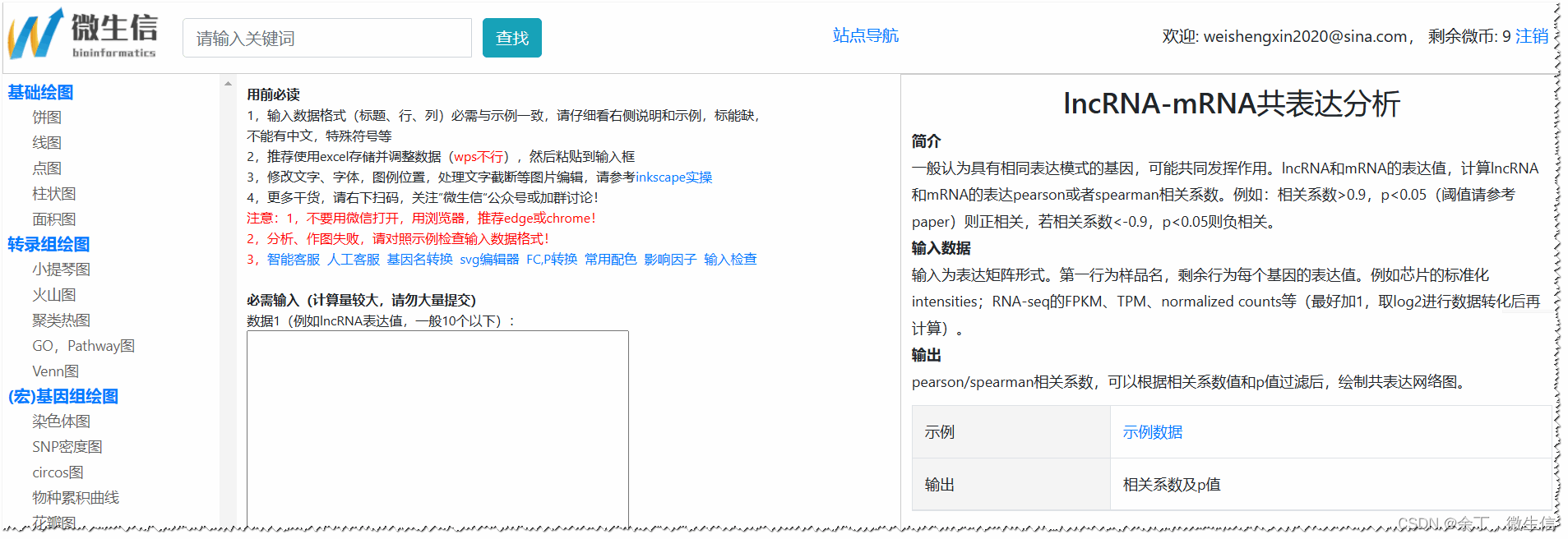
图3.相关系数计算页面
2,示例数据
点击右侧“示例数据”链接下载excel格式的示例数据。
示例数据(仅供参考)分两部分,上半部分是lncRNA表达量,下半部分是mRNA表达量。其中行是基因,列是样品名。LncRNA和mRNA的样品名顺序必需保持一致。

图4.输入数据
注意:需要参考示例数据,在excel中将自己的数据整理成示例数据的样式,每个cell都需要有数据,不能有空的单元格。
3,粘贴示例数据
拷贝示例数据中上半部分的lncRNA数据,粘贴到第一个输入框。拷贝示例数据中下半部分的mRNA数据,粘贴到第二个输入框。
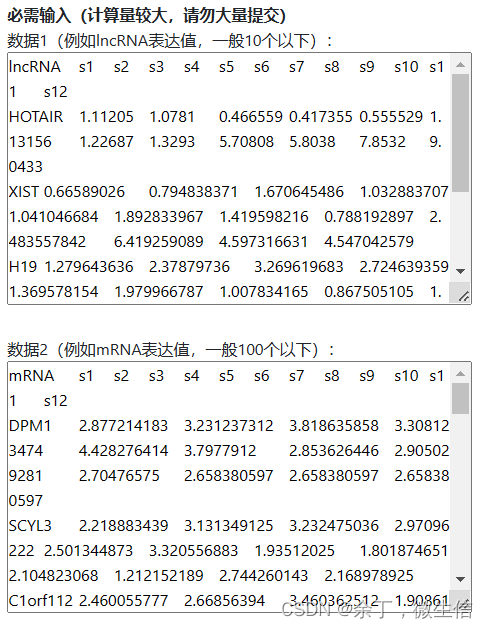
图5. 将数据粘贴到输入框
注意:不是拷贝excel文件,是拷贝excel文件里边的数据。另外粘贴到输入框后,格式乱了没关系,只要在excel中是整齐的就行。并且数据矩阵中不能有空的单元格,中文字符等。
4,修改参数,并提交
我们设置了数据是否转化、相关系数算法等参数。由于示例数据来自芯片,因此这里不转化。使用pearson相关系数进行计算。

图6. 可选参数
5,提交分析
粘贴好输入数据,调整好参数后,点击提交按钮,3秒钟后,会在页面右侧出现结果。

图7.结果说明及下载
结果以excel存储。

图8. 相关系数结果
各列说明:
LncRNA:lncRNA名字
mRNA:mRNA名字
r:pearson相关系数
pvalue:p值
flag:+:正相关,-:负相关
6. 过滤结果
下载结果后,使用excel的筛选功能进行过滤,这里以p<0.05,|r|>=0.4的lncRNA-mRNA对绘制共表达网络(带header共28行)。
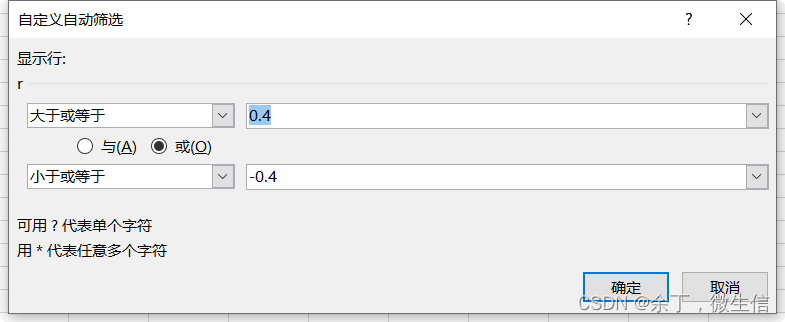
图9. 相关系数过滤

图10. P值过滤
7,导入cytoscape
拷贝这28行数据,粘贴到一个txt文件中。然后打开cytoscape软件,导入这个txt文件。在弹出的窗口中选择source和target。
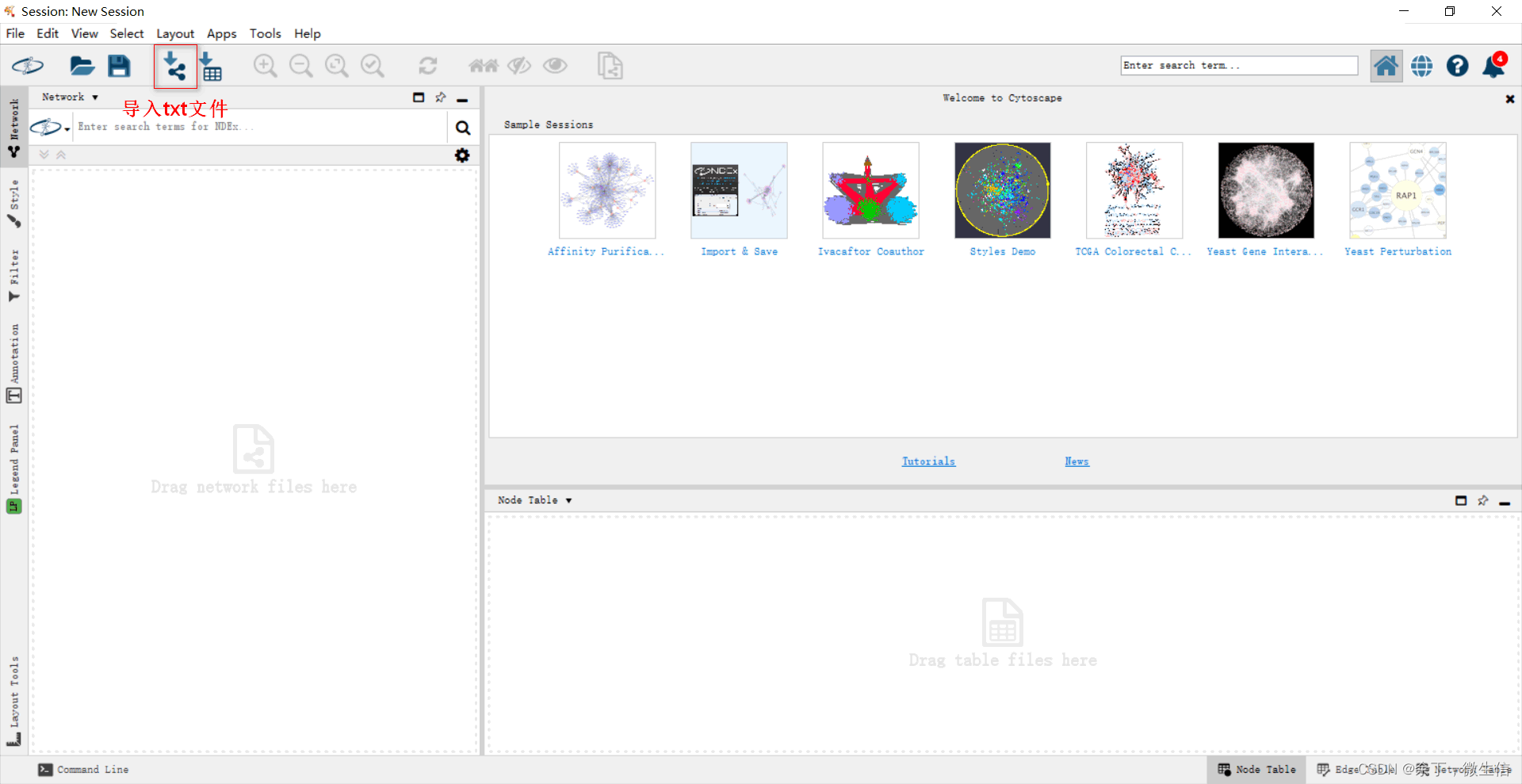
图11.导入txt

图12. 选择source和target,其中lncRNA为source,mRNA为target

图13. 默认网络图
8. 网络图美化
经过对颜色,节点形状,线型,布局等的简单美化后,获得最终的lncRNA-mRNA共表达网络图(图12)。由于每个人的审美不同,因此这里需要花费大量的时间进行美化,有些研究者还会使用AI等软件给网络图添加背景色等进行进一步优化。
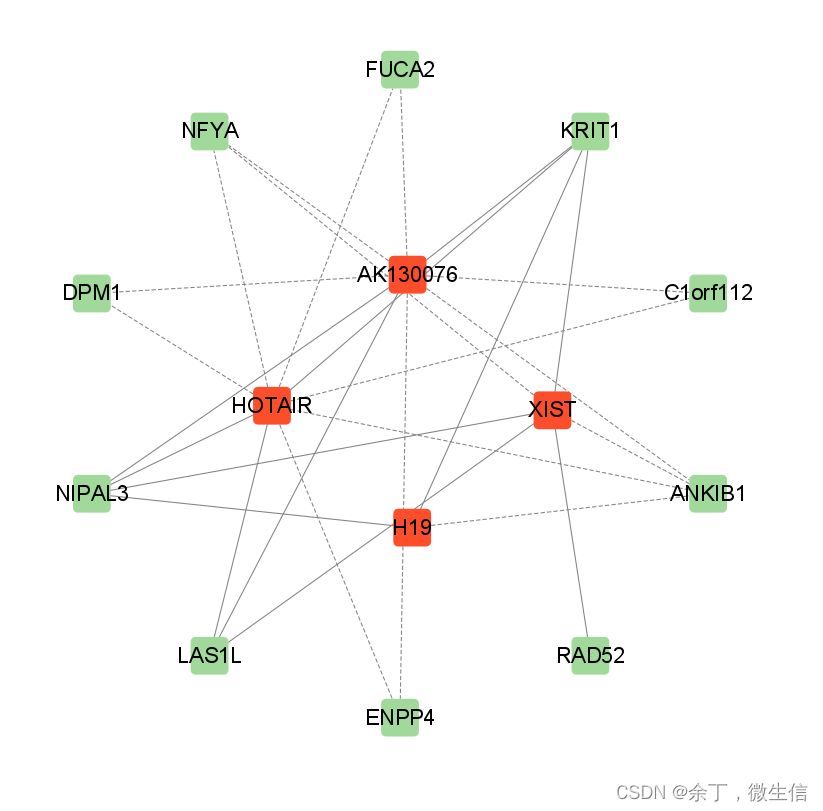
图14. 简单美化后的网络图
微生信助力高分文章,用户66000+,引用860+












![P8630 [蓝桥杯 2015 国 B] 密文搜索](https://img-blog.csdnimg.cn/8aa439044d284067956877bbb7e453a2.png)