摘要:氧化物负载的贵金属纳米粒子是广泛使用的工业催化剂。由于费用和稀有性,开发降低贵金属纳米颗粒尺寸并稳定分散物质的合成方案至关重要。负载型原子分散的单贵金属原子代表了最有效的金属利用几何结构,尽管由于合成均匀且稳定的单原子分散体的困难以及缺乏位点特异性表征方法而引起了关于负载型单贵原子种类催化活性的争论。我们提出了一种催化剂结构和表征方法来克服这些限制,通过在每个载体颗粒上沉积〜1个贵金属原子,并通过关联扫描透射电子显微镜成像和CO探针分子红外光谱来表征结构。这对于负载在锐钛矿TiO2上的Pt 得到证实。在这些结构中,孤立的 Pt 原子(Pt iso)在各种条件下保持稳定,光谱证据表明 Pt iso物种存在于均匀的局部环境中。将 Pt iso与 TiO2上约 1 nm 预氧化(Pt ox)和预还原(Pt金属)Pt 簇进行比较,我们识别出与每个位点结合的 CO 的独特光谱特征,并发现 CO 吸附能有序:Pt iso ≪ Pt金属< Ptox。Pt iso物种的 CO 氧化转换频率比 1 nm Pt金属簇高 2 倍,但具有相同的反应机制。我们提出活性催化位点是与TiO2键合的阳离子界面Pt原子,并且Pt iso表现出最佳的反应性,因为每个原子都暴露于催化作用并与TiO2形成界面位点。这种方法对于研究负载的贵金属原子的行为通常很有用。
Introduction
负载型铂族金属催化剂因其在广泛的化学转换技术中的应用而具有至关重要的重要性,其中对金属质量需求最高的是汽车三元催化转化器。由于铂族金属在地壳中的天然稀缺性,大量努力已经被投入到开发最大化金属利用效率的方法中,从而降低成本并促进可持续性。最大化金属利用效率的主要方法是通过减小纳米级粒子的大小来增加金属表面积与体积比率,使得更大比例的总金属原子在催化剂中可用于推动化学反应。在极限情况下,这对应于单个金属原子沉积在载体上,这类材料被称为单原子、原子分散或孤立位点催化剂,它们展现了完美的金属利用率,其中所有原子都暴露给反应物并可用于推动催化反应。推动孤立位点铂族金属催化剂发展的关键考虑因素是这些材料是否将保持或增强小纳米粒子的固有反应性,以实现特定活性的提高和催化剂成本的降低。对孤立位点铂族金属催化剂的额外兴趣还源于这样一个想法:这些材料可能会弥合非均相催化和均相催化之间的差距,潜在地提供在困难的化学转换中调节选择性的独特机会。
对于孤立的Pt族金属原子以及更广泛的贵金属原子在氧化物载体上的研究已经持续了几十年,最初的研究重点在于提供它们存在的光谱学证据。例如,通过使用红外光谱学将Rh(CO)2指认为与氧化物上孤立Rh原子结合的CO之后,研究工作开始致力于识别这些物种的结构、形成机制和局部几何结构。尽管最初的研究集中在通过光谱学识别氧化物载体上孤立的贵金属原子,但通过像差校正扫描透射电子显微术(STEM)直接观察载体上单个异质原子的能力,激发了对孤立位点贵金属催化剂的合成、表征及其潜在独特反应性的新兴兴趣。针对孤立位点贵金属催化剂在包括水煤气变换、二氧化碳加氢、乙烯二聚、丁二烯加氢和一氧化氮还原等反应中的反应性,文献报告日渐增多,这表明了一系列广泛且令人兴奋的潜在应用领域。一个受到显著关注的系统是氧化物载体下孤立Pt原子(Pt_iso)对CO的吸附和氧化作用。这种关注源自于反应的表面简单性,更重要的是,利用Pt_iso物种有可能减少汽车(主要是柴油发动机)CO氧化催化中对Pt的巨大需求。有趣的是,报告中CO与氧化物载体的Pt_iso之间的相互作用强度以及这些物种在CO氧化反应中的活性存在显著差异。例如,有理论和实验报告指出,与金属Pt团簇(Pt_metal)表面相比,CO与Pt_iso物种的结合力更弱或更强,其结合能根据载体和分析方法的不同,范围从20到>200 kJ/mol。同样,关于Pt_iso与Pt_metal团簇在CO氧化反应中的相对反应性也报告存在差异,其中Pt_iso物种被认为是更活跃或较不活跃的活性位点。
在氧化物负载的Pt单体物种(Pt_iso)上,CO的相互作用强度和反应活性的变化显然与特定的金属氧化物载体组成有关,这种组成作为配体修改了Pt_iso物种的反应活性。然而,鉴于类似类别的载体(例如,可还原的,如CeO2、FeOx、TiO2)之间的结论范围与不一致性,CO与Pt_iso之间的相互作用强度和报告活性的变化很可能也与在氧化物载体上合成稳定的Pt_iso物种以及这些物种的明确鉴定所关联的挑战有关。虽然已经采用了不同的方法将Pt_iso物种沉积在氧化物载体上,但一致的报告是,在高温氧化(煅烧)、还原或暴露于反应条件期间,Pt_iso倾向于聚集成小的Pt簇。由于Pt_iso在用于激活Pt催化剂的标准处理反应中稳定性的缺乏,使得严格鉴定Pt_iso的反应活性变得困难。例如,在高温煅烧条件下稳定性的缺乏,需要采用温和的氧化处理,这导致在催化剂上保留了可能影响Pt复合物的反应活性和迁移性的配体或反离子。此外,Pt_iso物种不能在升高的温度下暴露于H2,就可能存在与Pt_iso物种难以区分的小型氧化Pt(Pt_ox)团簇,鉴于这些结构中Pt的类似阳离子电荷和局部键合环境,很难将其与Pt_iso物种区分开来。最终,反应条件下的稳定性不足使得从Pt金属团簇中分离出Pt_iso物种的反应性变得困难。为了严格分析负载Pt_iso物种的反应性,关键是开发策略,在支持的Pt催化剂的标准预处理条件下保持其结构。
除了合成稳定的Ptiso物种外,还需采用严格的样品平均及位点特异性表征手段,以区分Ptiso、Ptmetal和Ptox。虽然STEM成像提供了单原子存在的直观证据,但由于该技术的统计局限性,以及无法从二维(2D)投影中提供载体表面的三维(3D)结构表示,这表明STEM应与其他补充表征技术结合使用。如X射线吸收(XAS)和X射线光电子谱(XPS)等基于光子的光谱技术,已广泛用于识别支撑金属催化剂的电荷状态和局部配位环境。这些技术在区分Ptiso物种的特征(如阳离子电荷状态或仅与氧原子配位)时受限,因为这些也是小型负载型Ptox的特征。由于这些相似点,通过XPS或XAS区分Ptiso与Ptox需要在Ptox簇还原为Ptmetal的条件下保持阳离子电荷且没有Pt-Pt配位。此外,样品中即便存在极少数的Pt团簇,也会导致结果不明确,因为在XAS中计算配位数时会对所有Pt物种的信号进行平均处理。
探针分子红外(IR)光谱法与CO是一种广泛使用的表征技术,它允许探测贵金属负载的局部结构、氧化态和配位环境。CO探针分子IR是一种样品平均技术,由于CO吸附到不同类型的贵金属位点时的振动频率不同,它也是具有位点特异性的,可以以温度程序方式运行,以提取有关不同贵金属位点化学反应性的信息。与XAS或XPS的问题类似,区分CO吸附到Ptiso和Ptox团簇时的振动带需要考虑,因为之前关于这些物种的报道确定它们的伸展频率都存在于∼2080-2130 cm–1的范围内。CO吸附到Ptiso和Ptox时的振动频率相似,比Ptmetal上的CO伸展频率(2030-2100 cm–1)高,这是由于这两种结构中Pt的阳离子电荷相似所致。
在所有表征方法中一致地看到,需要开发合成方法来在氧化物载体上创建稳定的 Ptiso 物质,这些物质可以在煅烧、还原和反应条件下存活,从而明确地区分 Ptiso、Ptox 簇和 Ptmetal 簇。区分这些物种的重要性得到了报告的证实,该报告显示,Ptox 物种(从含有几个 Pt 原子的小簇到扩展的单晶表面)表现出与 CO 的强烈相互作用和最小的 CO 氧化反应性,如果这些物种可能会混淆 Ptiso 反应性的测量没有区别和明显比较。
在这里,我们介绍了一种合成稳定 Ptiso 催化剂的通用方法,即使用直径约 5 nm 的氧化物纳米颗粒(本例中为 TiO2)作为载体,并控制 Pt 重量负载和合成条件,以在每个载体颗粒上沉积约 1 个 Pt 原子。通过使用相关的 STEM 和 CO 探针分子 IR 表征方法,结果表明,经过高温 (450 °C) 煅烧、H2还原 (300 °C)、暴露于 CO 氧化,Pt 在 TiO2 颗粒上保持位点隔离反应条件为〜3天,并在架子上老化〜1个月。 Ptiso 上的 CO 吸附具有以 2112 cm-1 为中心的尖锐(6–8 cm-1 半峰全宽,fwhm)IR 谱带,不会改变频率、宽度或覆盖范围的对称性,而 CO 吸附在 Ptiso 上直径为 1 nm 的 Ptmetal (2040–2100 cm-1) 和 Ptox (2100–2125 cm-1) 簇的特点是频率和形状随覆盖范围变化的较宽谱带。程序升温分析表明,CO 和 Pt 物质之间的相互作用强度顺序为 Ptiso < Ptmetal < Ptox。在没有传热和传质效应的稳态 CO 氧化测量中比较了 Ptiso 和 Ptmetal 的反应性,结果表明,在 200 °C 下,与 1 nm Ptmetal簇相比,Ptiso 的周转频率 (TOF) 高出 2 倍,但在相同的反应机理。我们根据 TiO2 负载的 Pt 上的 CO 氧化活性位点(界面阳离子 Pt 原子)来解释结果,证明当负载在 TiO2 上时,Ptiso物质在单位质量和位点的基础上表现出最佳的反应性,因为每个原子都暴露于催化和用载体创建界面位点。预计这里演示的方法通常可用于对各种氧化物上分离的贵金属的光谱和反应行为进行严格的分配。
2.实验方法
2.1材料
高纯度 (99.995%) 硝酸四氨铂 (II) (TAPN) 购自 Sigma-Aldrich(编号 482293)并用作 Pt 前体。具有高表面积 (290 m2/g) 的五纳米直径锐钛矿型 TiO2 (99.5%) 晶体购自 US Research Nanomaterials(库存号 US3838),并用作本研究的载体。催化剂合成中使用的试剂级 NH4OH(28-30% 浓度)购自 Acros Organics(编号 423300250)。 SiO2 凝胶(Davisil 等级 62,孔径 150 Å,60–200 目,部件号 243981,Sigma-Aldrich)和酸纯化 SiO2(Sigma-Aldrich,编号 84880)分别用于催化剂共浸渍和催化剂稀释。对于红外和稳态反应实验,使用以下气体:10% H2/Ar(±2%,空气)、He(UHP,99.999%,空气)、10% O2/He(±2%,空气) ) 和 10% CO/He (±2%, 空气气体) 在铝罐中,以防止金属羰基化合物进入反应物流。
2.2 Catalyst Synthesis
通过称为强静电吸附 (SEA) 的湿浸渍技术合成了以 Ptiso为主的催化剂,使用低 Pt 重量负载 (0.025–0.15 wt%)、大合成体积以促进 Pt 在载体上的均匀沉积,并控制溶液 pH 值 (8–12.5)。在仅含有 Pt同种物质的 0.025 wt% 催化剂的典型合成中,将 1 g TiO2粉碎,筛分至小于 75 μm 的粉末,并在合成前在真空烘箱中于 120 °C 干燥过夜。将25毫升去离子水与75毫升NH4OH混合以溶解TiO2载体,产生12.2的溶液pH。单独地,将10mg TAPN溶解在5mL去离子水中,从中取出300μL并添加到25mL NH4OH中以产生pH为12.2的前体溶液。将25mL TAPN溶液在12.5小时内注入支持溶液中,同时不断搅拌以实现125mL的最终总溶液体积和2900m2/L的表面负载(参见等式1 ) 。缓慢添加前体后,将最终溶液加热至 70°C 直至完全干燥。

为了合成含有 Pt 簇且不含 Ptiso的 1 wt% Pt 催化剂,使用了初湿(干)浸渍 (DI) 程序。在1克催化剂的典型合成中,将20毫克TAPN溶解在1200微升去离子水中,并将该溶液添加到990毫克TiO2(如上所述干燥和筛分)中,形成糊剂。将糊状物加热至70°C直至完全干燥。将干燥后的催化剂置于120℃真空烘箱中过夜并过筛至75μm。
所有催化剂均在管式炉中以 10°C/min 的速率升温后在流动空气中于 450°C 下煅烧 4 小时。高温和延长的煅烧时间确保从 TAPN 前体中除去所有剩余的胺或硝酸盐配体,这些配体在这些条件下完全分解。
2.3 STEM Iamging
STEM 成像是在配备两个球面像差校正器和 300 kV 冷场发射枪的 JEOL JEM-ARM300F Grand ARM 透射电子显微镜上进行的。使用 22 mrad 的会聚半角以及分别为 83 和 165 mrad 的内收集角和外收集角记录高角度环形暗场 (HAADF)-STEM 图像。每个样品收集 20-30 张图像,以观察至少 100 个 Pt 物种,并实现 CO 红外光谱的令人信服的结构分配。设置 STEM 成像条件以尽量减少电子束效应。使用相对较小的探针电流 20 pA 进行成像。同时,放大倍数始终保持在8000000以下,采集时间小于16秒。观察到单个 Pt 原子在这些条件下是稳定的。
2.4 IR Characterization
在收集 CO 探针分子红外光谱之前,将催化剂装入 Harrick 高温反应室(ZnSe 窗口)中,该反应室安装在 Nicolet iS10 FTIR 光谱仪中的 ThermoScientific Praying Mantis 漫反射适配器内,并配有由碲化汞 (MCT) 检测器冷却的液氮。使用质量流量控制器 (Teledyne Hastings) 控制穿过反应器床层的气体流速,所有气体首先通过保持在 -80 °C 的异丙醇液氮冷阱和填充有 Drierite 干燥剂的玻璃阱以去除微量水分。由于Harrick反应器床中的已知梯度,使用光学高温计校准催化剂床中的表面温度。
CO 探针分子红外光谱用于识别吸附在各种 Pt 结构上的 CO 的独特振动特征。为了检查每个结构上CO的光谱特征,以及O2流中CO的稳定性和反应性,使用了以下方案。首先,进行原位氧化(在 10% O2/He 混合物中,50 sccm,300 °C,30 分钟)或还原(在纯 H2中,240 °C ,50 sccm,1 小时)预处理,以允许区分来自 Pt金属簇和 Pt iso物种的Pt ox簇。接下来,将催化剂在以100sccm流动的He中冷却至室温。一旦达到室温,通过以 50 sccm 流动 10% CO/He 10 分钟,CO 被吸附直至饱和覆盖,其中观察到与 Pt 上 CO 吸附相关的谱带不再改变。该系统在 He 中以 100 sccm 冲洗 2 分钟,以去除任何气相 CO,其振动带与阳离子 Pt 物质上吸附的 CO 相关带重叠。接下来,在程序升温解吸(TPD)实验或程序升温氧化(TPO)实验中探讨了剩余化学吸附CO的稳定性。在 TPD 实验中,在 100 sccm 的 He 流中,温度以 ∼7 °C/min 的速率从室温升至 500 °C。或者,在 TPO 实验中,将氧气(50 sccm 的 10% O2/He)引入仍保持在室温下的化学吸附 CO,然后以 10 °C/min 的升温速率升温至 500 °C。在整个实验过程中,光谱均以 Kubelka-Munk (KM) 单位记录,扫描 32 次,数据间距平均为 0.482 cm–1光谱,每次测量之间允许半分钟增量。
2.5 Steady State CO Oxidation Kinetics
在玻璃管式填充床反应器中进行差动反应器测量,以直接测量和比较各种制备的催化剂的稳态CO氧化反应性。在所有情况下,均使用半英寸玻璃管,催化剂床的长径比为 4:1,以尽量减少沟流的可能性。将煅烧的Pt/TiO2催化剂筛分至<75μm,并以催化剂与SiO2的1:4比例与惰性SiO2凝胶共浸渍。将混合物在圆底烧瓶中与水一起超声处理 30 分钟,并搅拌过夜。然后将烧瓶转移至旋转蒸发器并以110rpm真空干燥30分钟。然后将整批共浸渍催化剂进一步用酸纯化的SiO2 (Sigma-Aldrich,编号84880)稀释5:1并装入填充床反应器中。使用稀释和共浸渍步骤来最小化(1)气流通过 5 nm 直径 TiO2颗粒填充床时产生的压降,(2)包含 5 nm TiO2颗粒的域的尺寸,这将引入传质和扩散对动力学速率的影响,以及(3)与这种高度放热反应相关的热点形成。催化剂共浸渍后,没有观察到催化剂床的压降,并且在所有感兴趣的流速下,反应速率保持恒定,表明没有传质限制(即,动力学测量是在反应限制的情况下进行的) 。
对于稳态 CO 氧化测量,将 50 mg 催化剂装入反应器中,并在 20 sccm 和 300 °C 下用纯 H 2还原 2 小时进行预处理。将催化剂在160 sccm He中冷却至200℃,然后暴露于反应条件(1%CO、1%O2、98%He,总流量200sccm)10小时,以达到稳态;在 10 小时内,反应速率变化小于 5%。活化期后,将反应器以相同的组合物冷却至 140°C,并在 2 分钟内以 10°C 的温度增量进行活化势垒测量,然后在给定温度下保持 2 小时,以确保稳定在质谱仪 (MS) 中达到状态信号。分压相关测量是利用自制的可编程 Arduino 控制器进行的,该控制器自动化电磁阀控制,允许反应物绕过反应器,在每次压力变化之前和之后在 MS 中进行基线测量。在进行分压相关测量之前,对活化屏障测量进行相同的预处理,包括在 200 °C 下运行 10 小时。激活期后,不进行冷却,而是在 200 °C 下执行分压相关测量。所有分压测量的总流量在 He 平衡中为 200 sccm。首先运行CO分压依赖性,以1.5%CO与0.5%O 2的最高CO进料比开始,并以0.25%CO与0.5%O 2的最低CO进料比结束。在每个进料比开始和结束时,反应物绕过反应器流动 20 分钟,以在 MS 中获得基线信号。在基线测量之间,反应物流过催化剂床 2 小时以获得稳态信号。紧接着最低的CO进料成分,同时仍处于200℃,在40分钟的旁通期之后,以最高的O2进料比(0.5%CO比1.5%O2)开始O2分压依赖性测量。交替旁路/反应器循环继续进行7次稳态测量,其中O2进料组成依次降低且CO压力恒定,最终进料比为0.5% CO与0.25% O2。对于两组反应实验和所有催化剂,报告的动力学测量结果是至少3次独特测量结果的平均值(即,将新的催化剂制剂装入反应器中),以确保可重复性和一致性。使用可编程 PID 控制器 (Omega CN7800) 控制自制熔炉中的温度。流速由校准的质量流量控制器(Teledyne Hastings)控制。在反应器之前使用 Drierite 床清除气流中的湿气(添加异丙醇液氮捕集器的测试显示对速率没有影响),并购买了铝衬 CO 罐以防止金属羰基形成并确保清洁CO 流。所有排出气体均使用校准的在线质谱仪(Halo 201,Hiden Analytical)进行测量。
Results
3.1 Synthesis and Vibrational Band Assignments of CO Adsorbed to Ptiso
据推测,通过使用小型氧化物纳米颗粒作为载体并在每个载体颗粒上沉积约 1 个 Pt 原子,可以制备具有高稳定性的 Ptiso物种。在这种几何结构中,即使 Pt 原子在氧化物表面上移动,载体颗粒之间所需的迁移也应最大限度地减少形成 Pt 多聚体的团聚。为了合成这种结构,我们使用了约5 nm直径的锐钛矿TiO2颗粒(表面积为290 m2 /g)和低Pt重量负载,相当于每个TiO2颗粒少于1个Pt原子(0.025-0.05 wt%相当于0.2-0.4 Pt 原子/TiO2颗粒)。如果将随机分布的 Pt 原子沉积到 TiO2颗粒上,则将有很大一部分 TiO2颗粒含有 >1 个 Pt 原子,请参见图1 A。为了减轻随机基础上预期的 Pt 原子聚集为了促进 Pt 原子在载体颗粒上的均匀沉积,大合成体积与长时间添加 Pt 前体相结合,形成均匀混合的稀释浆料。采用强静电吸附(SEA)湿浸渍技术的原理来促进溶液中Pt离子之间的排斥相互作用以及Pt离子与TiO2表面之间的吸引相互作用。通过添加NH4OH调节pH来修饰TiO2载体表面,羟基被去质子化,在TiO2表面形成O-阴离子,库仑吸引[(NH3) 4Pt]+2,如图1所示A. 通过小 TiO2颗粒、低 Pt 重量负载、均匀混合溶液和受控 TiO2 -Pt 阳离子相互作用的组合,预计很有可能产生稳定的 Ptiso物质。
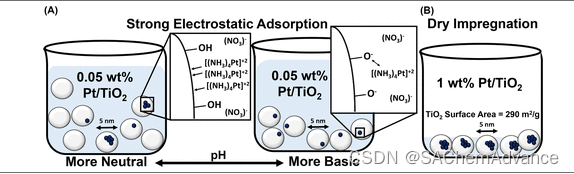
作为 Pt iso催化剂设计合成的比较,通过初湿(干)浸渍 (DI) 制备了 1 wt% Pt/TiO2催化剂,目的是生成 Pt团簇(图1 B): 1 wt%负载表明,平均每个 TiO2颗粒将沉积约 8.4 个 Pt 原子,溶液体积较小且缺乏 pH 调节,从而消除了 Ptiso形成的预期驱动力。 由于颗粒烧结所需的 TiO2 颗粒之间的迁移,以及每个 TiO2 颗粒沉积的铂原子数相对较少,即使在此合成过程中缺乏控制铂颗粒大小的具体工作,预计也会形成纳米和亚纳米直径的颗粒。
由于其对局部键合环境高度敏感,CO 探针分子红外光谱被用来分析合成参数如何影响所得的 Pt 结构。使用 CO 作为探针分子并通过红外表征负载型 Pt 催化剂的结构性质和电荷的文献很多。对于负载的 Pt金属颗粒,吸附 CO 的振动频率有明确的值。当CO吸附到Pt金属位点时,随着Pt-Pt配位数从∼5(Pt颗粒上的角或缺陷位点的特征)增加到9(扩展的特征(111),振动频率从2040增加到2100 cm–1)Pt 颗粒表面)。相反,当 CO 吸附到氧化物负载的阳离子 Pt 物质(Ptox簇、Ptiso物质或与氧化配体配位的 Pt)时,CO 振动频率从 Pt金属吸附位点蓝移,通常为 >2100 cm–1。与 Pt金属位点相比,吸附到阳离子 Pt 位点的 CO 的蓝移频率源自向 CO 的电荷转移减少以及 CO 键距离更接近气相 CO 距离。我们假设,对于使用上述 SEA 方法合成的催化剂,经过严格的煅烧(空气中 450 °C 4 小时)和原位还原(H2 中 240 °C 1 小时)进行预处理,任何剩余的 IR 谱带CO 吸附到拉伸频率 >2100 cm–1的 Pt 位点上一定是由于 Pt同种物种的存在,因为合成中的所有氧化配体都将通过煅烧除去,并且任何 Ptox簇都会在煅烧过程中还原为 Ptmetal簇。
图2 A 显示了在 pH 为 12、Pt 前体添加速率为 0.025 mg Pt 每小时以及不同的表面负载(与合成成反比)下合成的 0.05 wt% SEA 催化剂在饱和覆盖和室温下吸附 CO 的红外光谱溶液体积,或定性,稀释度)。将光谱归一化为 2112cm–1处 CO 伸缩带的强度,以比较吸附到 Pt金属位点 (2040-2090cm –1 )的 CO 相对量。可以看出,随着表面负载的减少(更稀释的合成条件),与 CO 在阳离子 Pt 位点 (2112cm–1 ) 吸附相关的谱带强度相对于与 CO 相关的宽带强度增加Pt金属位点上的吸附。
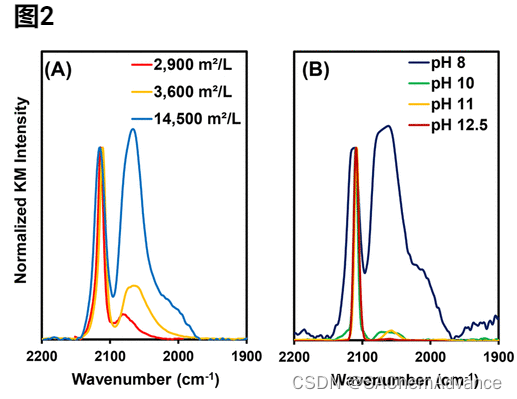
图2B显示了在 0.05 wt% SEA 催化剂的饱和覆盖度下吸附 CO 的光谱,该催化剂是在 Pt 前驱体添加速率为 0.025 mg Pt 每小时、表面负载为 3600 m2 /L 以及在 8 和 12.5 之间变化的 pH 值下合成的。
据观察,随着合成 pH 值的增加,催化剂倾向于包含单一 CO 吸附位点,其对称 CO 伸缩带的频率为 2112 cm–1且 fwhm <10 cm–1,并且优化的合成条件始终能够产生fwhm 为 6-8 cm–1。在接近中性 pH 值下合成的催化剂含有 Pt金属CO 吸附位点和以 2112 cm–1为中心的加宽不对称谱带,如图2 B所示。
优化条件为Pt前驱体添加速率为0.025 mg Pt/h、表面负载量为3600 m2/L、pH为12.2、Pt负载量为0.025-0.5 wt%,制备了一系列Pt/TiO2催化剂。 。该系列催化剂在饱和覆盖度下吸附CO的红外光谱如图S2所示。可以看出,当合成过程中标称少于 1 个 Pt 原子/TiO2颗粒(0.025-0.05 wt%)时,观察到频率为 2112 cm–1的单一、尖锐的 CO 谱带。当 Pt 重量负载增加到 1 Pt 原子/TiO2颗粒以上 (>0.1 wt%) 时,第二个宽 CO 带形成,频率为 2040–2090 cm–1,表明 CO 吸附到 Pt金属位点。随着 Pt 质量负载的增加, 2040-2090 cm–1处的谱带强度比相对于 2112 cm–1谱带增加。
为了总结合成方案对 CO 探针分子红外光谱的影响,发现当 Pt在高度稀释条件、高 pH 和重量负载低于 1 Pt 原子/TiO 的条件下沉积到 5 nm 锐钛矿 TiO2颗粒上时粒子时,观察到频率为 2112 cm–1且 fwhm 为 6–10 cm–1的单个 CO 伸缩带。考虑到在所有情况下催化剂均在已知将Pt氧簇还原成Pt金属簇的条件下用H2预处理,保留了CO吸附位点的存在,该CO吸附位点具有阳离子Pt原子上吸附的伸缩频率特征,并且CO吸附位点的尖锐度CO 谱带表明,将 2112 cm–1振动模式分配给吸附在锐钛矿 TiO2载体上的 Pt同种物质上的 CO 。
为了支持锐钛矿 TiO2上 Pt iso吸附的 CO 的红外分配,进行了相关的 IR-STEM 分析。我们重点研究了 3 个样品:(1) 通过优化的 SEA 方案合成的 0.05 wt% 催化剂,(2) 通过相同的 SEA 方案合成的 0.15 wt% 催化剂,以及 (3) 通过 DI 方法合成的 1 wt% 催化剂。两种SEA催化剂均使用12.2的pH、2900m 2 /L的表面负载和每小时0.025mg Pt的Pt前体添加速率来制备。对于这些样品,我们收集了饱和覆盖范围内的 CO 探针分子红外光谱,见图3 (图 A-C),以及相同材料的 20-30 张 STEM 图像,其示例如图3(图 D-F)所示,在进行两种表征之前,通过相同的方案对它们进行煅烧和还原。在 0.05 wt% SEA 催化剂的约 30 个 STEM 图像中,Ptiso是主要观察到的 Pt 物质,仅观察到约3 个直径 <1 nm 的 Pt 簇,图3D。观察到 Pt 的平均直径为 0.176 nm Ptiso物种,见图S3。Pt同种物质的测量直径不能与Pt原子半径直接相关,而是表明Pt同种物质的测量尺寸大于仪器的分辨率,因此可以将Pt同种物质与二聚体或三聚体区分开铂。识别 Pt同种物种的STEM 图像与同一 CO 饱和样品的红外光谱非常一致,图3 A,其中 2112 cm–1处的谱带强度比在 2112 cm–1处观察到的宽带强约 180 倍。 2040 和 2090 cm–1。对于 0.15 wt% SEA 催化剂,STEM 成像显示与 0.05 wt% SEA 催化剂相比,Pt 簇的数量(平均直径为 0.84 nm)显着增加,并且 Pt 同种物质的维持存在,图3 E 非常吻合图3 B是 CO 饱和催化剂的红外光谱,显示与 0.05 wt% SEA 催化剂相比, 2040-2090cm–1处 CO 伸缩带的相对强度有所增加。最后,1 wt% DI 催化剂的 STEM 图像显示主要存在平均直径为 1.1 nm 的簇和一些平均直径为 4.3 nm 的较大颗粒(图3F),证实了 CO 饱和催化剂的 IR图3C,几乎完全显示 2040 至 2090cm–1之间的 CO 伸缩带强度。
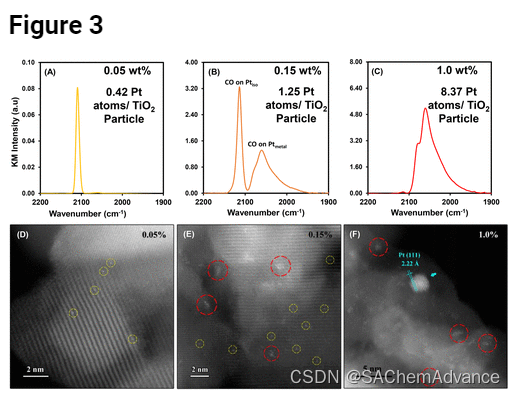
STEM 成像显示了确凿的证据,表明在煅烧和还原后,0.05 wt% SEA 催化剂上主要保留了 Ptiso物质,正如合成方法设计中所假设的那样。此外,相关的STEM-IR分析提供了令人信服的证据,表明2112 cm–1处的尖锐CO伸缩带可以归因于锐钛矿TiO2上的Ptiso上吸附的CO 。
3.2 Site-Specific Signatures of CO Adsorbed to Ptiso, Ptmetal, and Ptox
由于经过还原处理后 Pt同种物质在 5 nm TiO2颗粒上表现出稳定性,我们选择了 1 wt% DI(主要含有 Pt 簇)和 0.05 wt% SEA(主要含有 Pt同种物质)催化剂来识别区分不同物质的光谱特征CO 吸附在 Ptiso和 Ptox簇上,它们都含有阳离子 Pt 原子。为了实现这一目标,两种催化剂首先在流动的 H2中于 240 °C 预处理 1 小时,然后在室温下暴露于 CO 至饱和覆盖并通过红外表征。然后将催化剂氧化以去除吸附的CO并氧化Pt结构(280°C,在10% O2中30分钟),冷却至室温,并再次暴露于CO以通过红外表征。通过在相同的催化剂上依次进行实验,可以将吸附到 Ptmetal和 Ptox簇的 CO 光谱与吸附到已预氧化或预还原的Pt同种物质上的 CO 进行比较。
在图4 A 中可以看出,还原后,1 wt% DI 催化剂在饱和覆盖度下吸附的 CO 光谱显示,在与以2077cm-1 为中心的Pt金属位点吸附的 CO 相关的区域中,两个主要的 CO 拉伸带重叠,分配给良好配位(WC)Pt 位点,2058cm-1,分配给欠配位(UC)Pt 位点。光谱显示在与吸附到阳离子 Pt 位点 (>2100cm-1 ) 的 CO 相关的频率范围内几乎没有强度,表明完全还原的 Pt 簇。

1 wt % DI 催化剂氧化后,与还原催化剂相比,吸附 CO 的光谱显示与 CO 结合到 Pt金属位点 (2040-2090 cm–1 )相关的谱带强度降低,并且出现与吸附到阳离子 Pt 位点 (>2100cm–1)的 CO 相关的宽带,参见图4 A。使用多个高斯峰的最小二乘拟合进行峰解卷积表明,与阳离子 Pt 位点吸附相关的 CO 伸缩带位于中心约为 2118cm–1,半峰宽为 25-30cm–1,图 S4。还原和氧化处理后的 1% DI 催化剂的 STEM 成像显示出基本相同的 Pt 结构几何形状,主要包含平均直径为约1 nm 的 Pt 团簇和一些平均直径为约4 nm 的较大 Pt 颗粒,图S5。鉴于氧化和还原处理后 Pt 颗粒缺乏结构变化,图4 A中约2090-2140cm–1之间的宽 CO 伸缩带被分配给 Pt ox簇上的 CO 吸附,与之前的报道一致。图4 A中在氧化处理后观察到的频率低于 2090cm–1的 CO 伸缩带被分配给被流动的 CO 还原的 Pt 结构(或 Pt 结构的一部分),正如之前在 Pt/TiO 2催化剂中观察到的那样。然而,综合IR和STEM数据显示,氧化和还原处理后Pt没有显着的结构变化,很明显,CO 仅部分还原了 Ptox簇。这是讨论部分将要讨论的重要一点,因为氧化 Pt 团簇被 CO 唯一部分还原表明除了保留的氧之外还可能存在含有还原 Pt 金属物质的 Pt 结构,并且通过 CO 的观察得到了支持在与金属和阳离子 Pt 原子吸附相关的频率上拉伸能带。
与图4 A中看到的 Pt 簇的行为相反,其中分配给阳离子 Pt 位点吸附的 CO 伸缩带仅在氧化预处理后出现,对于 0.05 wt% SEA 催化剂,观察到主要的 CO 伸缩带位于 2112 cm–1,我们将其指定为吸附在 Ptiso上的 CO ,在还原性气氛中预处理后出现,参见图4 B。然而,在催化剂预氧化和 CO 吸附之后,红外中 CO 伸缩带的整体强度光谱显着下降(100 倍),导致 2040-2090 cm–1和 2090-2130 cm–1的谱带,与图4 A中所示的预氧化 1 wt % DI 的光谱相似。同种物质要么表现出与CO相比对O2的优先吸附,要么TiO2晶格中的空位引起迁移,从而使Pt同种物质中毒。因此,氧化处理后,Pt同种物质中毒并且不允许 CO 吸附,而该催化剂中的少量 Pt 簇以还原原子和阳离子原子的组合存在,类似于 1 wt% DI 催化剂。
简而言之,通过使用分别含有 Pt 簇和 Pt 同种异构体的 1 wt% DI 和 0.05 wt% SEA 催化剂,并控制氧化或还原预处理,我们可以区分吸附到 Pt iso上的 CO 的红外光谱。 、Pt ox团簇和 Pt金属团簇。这是至关重要的,因为两种阳离子 Pt 物质上的 CO 吸附显示出具有相似拉伸频率的谱带(Ptiso上为 2112 cm–1 , Ptox簇上为2118 cm–1),主要区别在于谱带宽度(<10 cm–1在 Ptiso上,25-30 cm–1在 Ptox上),这使得它们难以区分。与 Ptiso、 Ptox和 Ptmetal位点上的 CO 吸附相关的振动带的严格分配使得能够通过红外中的 TPD 实验来比较每个吸附位点上的 CO 吸附强度和 CO 解吸过程的特征。
图 S6(图 A 和 B)显示了分别在已预还原和预氧化的 1 wt% DI 催化剂上进行的 TPD 实验(β = 7 °C/min)期间记录的与各种温度相关的红外光谱。在预还原催化剂上,观察到 CO 解吸在约50°C 时从 Pt金属位点开始,并在 300°C 时大部分完成[图 S6(A)]。解吸温度高于 Pt 团簇中 CO 的 TPD 实验中传统的解吸温度。我们将这种差异归因于平均给定光谱中收集的数据所需的时间,导致每个光谱代表一定的温度范围。随着温度的升高,由于 Pt金属表面上相邻 CO 分子的偶极-偶极相互作用减弱,导致能带强度降低,CO 伸缩频率发生特征性红移。对于预氧化的 1 wt % DI 催化剂,观察到与 Pt金属位点 (<2100cm–1 ) 相比, CO 与阳离子 Ptox位点 (>2100cm–1) 的结合更牢固,并且从 Ptox上显着解吸位点仅在 200 °C 以上出现,甚至在 350 °C 时也不完整[图 S6(B)]。与 Pt金属簇相比, CO 对 Pt氧化簇的吸附更强,这与之前在 Pt 单晶和负载型 Pt 上的结果一致。在 CO 从 Ptox位点解吸的过程中,观察到 CO 带频率和宽度的各种变化,表明在温度升高期间氧化态和吸附位点结构的演变。
与 CO 在 Pt 簇上表现出的稳定性相反,图5A显示了在室温下流动的 He 下,在还原的 0.05 wt% SEA 催化剂上,Pt 同位点被 CO 饱和后收集的红外光谱。据观察,如图 S7所示,按照一级单分子解吸动力学,所有 CO 在 30 分钟内从Pt 同位点解吸,因此没有执行升温。很明显,与 Ptmetal或 Ptox位点相比,CO 与 Pt同位点的结合更弱。除了在 Pt同位点上观察到的 CO 稳定性较低之外,在整个解吸过程中,CO 伸缩带保持对称,半峰宽保持恒定在 ∼8 cm–1。此外,当CO解吸时,振动频率是恒定的。CO 在 Pt同位点上的拉伸频率不存在依赖于覆盖范围的变化,这表明吸附的 CO 分子之间没有相互作用,因此 CO 必须在原子分散的 Pt 原子上空间隔离,这与在 Pt金属和Pt 上观察到的行为相反。Ptox位点见图S6。最后,解吸过程中 Ptiso上 CO 振动带保留的对称性和 FHWM有力地证明了 Pt-CO 物种本质上是均匀的,可能全部位于 TiO2载体上的共同吸附位点。
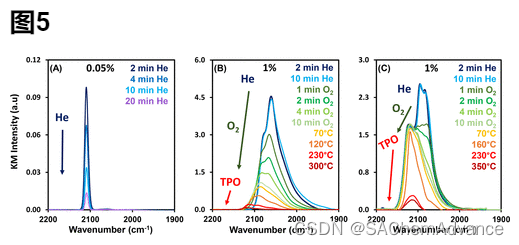
在了解 CO 与各个吸附位点相互作用的相对强度后,我们还想在瞬态实验中分析 CO 与这些位点结合的相对反应性。我们重点关注与 Ptmetal和 Ptox位点结合的 CO,因为 CO 与 Pt iso位点的结合太弱,无法分析瞬态实验中的反应性。图5(图 B 和 C)显示了在以下实验方案中分别在还原和氧化的 1 wt% DI 催化剂上收集的红外光谱:室温下 CO 吸附至饱和,室温下 He 暴露 10 分钟,10%O2 / He 混合物持续 10 分钟,并在 10% O 2混合物中进行程序升温氧化 (TPO) 升温 (β = 10 °C/min) 至 500 °C 。从图5B中可以看出,在室温下He存在下,吸附到Pt金属位点上的CO是稳定的,与图S6(A)一致。在室温下暴露于 10% O2/He 混合物时,CO 反应生成 CO2。(33)在 TPO 过程中,剩余的 CO 在 120 °C 时几乎完全转化为 CO2,只有少量 CO 吸附到 Ptox位点(在氧化斜坡期间形成),在 300 °C 以上之前保持完整。众所周知,这表明 CO 在 Pt金属位点上被 O2氧化的高反应活性。
相反,在图5C中观察到,与预氧化 1 wt% DI 催化剂上的Pt氧化位点结合的 CO 的反应性明显低于图5B中观察到的与 Pt金属位点结合的 CO的反应性,直到 >160 °C 为止反应性极小。如图4 A所示,最初,由于 Ptox簇被 CO还原,预氧化催化剂具有一些金属特性,正如 Ptmetal位点 (2040-2090 cm–1 )上吸附的 CO 伸缩频率特征所证明的那样。图5 C中突出显示了与 Ptox和 Pt金属位点结合的 CO 反应性的差异,在室温下引入O2后,与 Ptmetal位点结合的 CO 几乎完全损失,而与 Ptox结合的 CO 保留存在直到温度更高。
Ptiso、Ptmetal和 Ptox位点上的 TPD 和 TPO 实验突出了几个独特的特征,区分了吸附 CO 的位点特异性光谱和反应特性。首先,CO 吸附强度的顺序为 Ptiso ≪ Ptmetal< Ptox。其次,吸附到 Ptiso物种上的 CO 表现出恒定的 fwhm、带位置和带对称性,并且覆盖范围不断变化,这与 Pt ox和 Ptmetal位点上的观察结果形成鲜明对比,证明了产生的 Ptiso物种的均质性并进一步证实了IR 分配。第三,吸附到Ptox位点的CO在O2中的反应性明显低于吸附到Pt金属位点的CO,这表明Ptox位点在Pt/TiO2催化剂的稳态CO氧化反应性测量中不起任何作用。
关于CO反应活性的对比
具体参见论文原文
Discussion
与之前的报道相比,在氧化条件、还原条件、反应条件或 TEM 电子束下观察到 Pt 同种物质形成 Pt 簇,本文报道的 TiO2纳米粒子上的 Pt同种物质在以下条件下保留了其结构和均匀分布:所有考虑的条件。鉴于已知的 Pt 通过 Pt 原子发射和在载体上迁移的烧结机制,大多数 Pt同种物质在各种条件下缺乏稳定性并不令人惊讶。克服Pt同种物质的低稳定性的一种方法是提供锚定位点,例如配位不饱和Al 3+位点或CeO2上的方形平面结合袋,其中Pt同种物质结合特别强烈。然而,即使在这些情况下,也没有证据表明 Ptiso物种在还原条件下保持稳定,这是区分 Ptiso物种和 Ptox簇的反应性所必需的。
这里提出的另一种方法是在单个载体颗粒上分离单个 Pt 原子——有效地将 Ptiso物质束缚在TiO2岛上,这样 Pt 聚集就需要载体颗粒之间的能量上不利的跳跃。最近提出了这个想法,其中磷钨酸或磷钼酸已被用来承载单个 Pt 或 Rh 原子。这些小的酸性簇是单个金属原子的有趣载体,因为它们界限明确且很小。然而,这些催化剂的稳定性受到载体稳定性低的限制,载体在200℃以下会降解,从而限制了它们的应用。
这里报道的Pt同种物质在多种条件下的明显稳定性,并且载体上没有特定的锚定位点,这表明我们的合成策略成功地主要在每个 TiO2颗粒上沉积了单个 Pt 原子。在对图3D所示的 0.05% SEA 催化剂的 STEM 成像的分析中,包含 Pt同种物质的 30 张图像中只有 2 张显示出每个 TiO2颗粒存在多个 Pt同种物质的证据。即使在这些情况下,也不能排除图像中重叠的 TiO2颗粒是 Pt同种物种接近的来源。因此,STEM成像支持每个TiO2颗粒主要沉积1个Pt原子的假设。
这一结论与观察到的合成条件对 Pt同种物种与 Pt 团簇生产影响的趋势一致,支持了这一结论。例如,对于以与每个 TiO2颗粒少于 1 个 Pt 原子(<0.1 wt%) 一致的重量负载合成的催化剂,CO 在 Ptiso物质上的红外特征占主导地位,前提是合成是在极度稀释和均匀的条件下进行的。在混合条件下,溶液中 Pt 离子和 TiO2颗粒之间的相互作用很少,因此有利于每个 TiO2颗粒沉积单个 Pt 离子。此外,图S2中所示的重量负载相关测量表明,即使在使用优化的合成条件时,如果Pt重量负载指示平均每个TiO2颗粒应有大于1个Pt原子,则发生Pt簇形成。这些考虑因素共同提供了强有力的证据,证明我们的合成策略在足够低的重量负载下操作时,诱导每个 TiO2颗粒沉积约 1 个 Pt 原子,并且这种结构是 Ptiso物质表现出优异稳定性的主要原因。高稳定性对于区分 Ptiso物质与 Ptmetal和 Ptox簇的光谱特征和反应性至关重要。
接下来我们关注 CO 在各个 Pt 位点上的红外光谱和吸附能。使用红外和 TPD 对 Pt 表面、纳米颗粒和团簇上的 CO 吸附进行了详细研究,我们对 Ptmetal和 Ptox团簇的结果与之前的报道非常吻合。吸附到 1 wt% DI 催化剂上的1 nm Ptmetal簇上的 CO 表现出两个不同的谱带,分别以 2077 和 2058cm–1为中心,分配给簇上吸附到 WC 和 UC Pt 原子上的 CO。观察到与 UC 位点上的 CO 吸附相关的带强度更大,这与考虑到 STEM 观察到的 Pt 簇和粒径分布(直径为 1 nm 的簇,其中约 4纳米颗粒)。在预氧化的 1 wt% DI 催化剂上,吸附的 CO 的红外光谱在 2118 cm–1处显示出宽带,该谱带归因于 Ptox簇上的 CO 吸附。这一分配之前已进行过,并通过 STEM 分析得到证实,显示无论还原或氧化预处理如何,Pt 颗粒尺寸均恒定。与相同尺寸的 Pt金属簇相比,吸附在Ptox簇上的 CO 的蓝移伸缩频率源于Ptox簇中 Pt 的阳离子氧化态导致向 CO 的电荷转移减少。所有 IR 分析都与之前的结果一致。
TPD 观察到 CO 在 Pt氧簇上的吸附能比在 Pt金属簇上的吸附能更强,这由较高的解吸温度表明。这与之前的结果一致,在 TPD 实验期间,观察到 CO 从 Ptox解吸的最大速率发生在比 Ptmetal高 100 °C 的温度下(Ptox为280 °C ,Pt金属为 180 °C )。还原和氧化的 Pt 单晶。CO 在 Ptox簇上更强的吸附能解释了为什么这些位点对 CO 氧化表现出极低的反应性:CO 会毒害催化表面。这也凸显了区分 Ptiso和 Ptox物种在反应性分析中的重要性,因为如果不区分 CO 的大吸附能和 Ptox上的低 CO 氧化反应性可能会混淆 Ptiso位点的固有反应性。
虽然文献中对与 Ptmetal和 Ptox簇结合的 CO 进行红外光谱分析的文献很多,但对与氧化物支撑的 Pt同种物质结合的 CO 的严格分析却少得多。先前关于与 Pt iso物质结合的 CO 相关的指定频带的振动频率的报告随着载体的变化而变化(Pt iso /H-丝光沸石,2123 cm–1 ; Ptiso/H-ZSM-5,2115cm–1 ; Ptiso/CeO2 , 2095 cm–1 ;和 Ptiso/FeOx,2080 cm–1 ),但通常与此处报道的 Ptiso/TiO2的 2112 cm–1相当。CO 与 Ptiso物质结合的能带位置的变化可能是由 Pt iso物质与载体之间不同的相互作用引起的,之前的研究发现,当从酸性较强到酸性较强时,与负载金属结合的 CO 的伸缩频率会发生红移。所有报告中一致的是,与 Ptmetal簇相比,当与 Pt同种物质结合时,CO 伸缩频率蓝移约40cm–1。Ptiso物种上常见的蓝移 CO 伸缩频率源自阳离子氧化态(可能为2+),其源于 Pt iso与氧化物载体上的氧原子的直接配位。此外,与之前的一些报告类似,当吸附到 Ptiso/TiO2上时,与 CO 拉伸相关的谱带被观察到表现出作为 CO 覆盖范围的函数的恒定频率,这是由于缺乏偶极-偶极耦合而发生的相邻CO配体在分散吸附位点上的分布,是CO与孤立金属位点结合的关键光谱特征。
与氧化物载体上的单金属原子催化剂相关的两个新出现的重要问题如下。(1) 这些是单中心催化剂,即所有金属原子都位于载体上的同一位点上,还是金属原子分布在载体上的多种类型的位点上?(2) 贵金属原子位于载体上的什么位置?
有人提出,吸附的 CO 伸缩带的 fwhm 可用于定性解决金属原子在载体上分布的均匀程度问题,其想法是,较宽的 CO 带 fwhm 表明孤立的金属原子位于载体上。各种局部环境,从而在金属原子和 CO 之间产生一系列不同的相互作用。(68)之前关于 Pt iso物质吸附 CO 的红外光谱报告显示,fwhm 相对较宽,范围从 15 到 >30 cm–1(Ptiso/H-丝光沸石,∼18 厘米–1 ; Ptiso/H-ZSM-5,∼25 cm–1 ; Ptiso/CeO2 ,30 cm–1;和 Ptiso/FeOx,>30 cm–1 ),表明 Pt同种物质被吸附到载体上的一系列不同位点。相比之下,我们报告了当吸附到 Ptiso/TiO2时,CO 拉伸带的 fwhm 在 6 和 10 cm –1之间要紧得多,这表明 Pt iso物种均匀分布在单个或几个非常相似的位置, TiO2载体上的吸附位点。最近对精心制备的沸石载体上的 Ir 单原子进行的分析报告称,fwhm 约为 5 cm–1 ,与溶液中均质有机金属 Ir 羰基配合物的 4 cm–1 fwhm相比,这表明沸石负载的单 Ir 原子可能是单位点材料。将 Ir 情况与此处观察到的 Ptiso/TiO2吸附时观察到的 CO 拉伸6 cm–1fwhm 进行比较,我们的合成方法似乎能够将 Ptiso物种定位在 TiO2上的单个吸附位点,尽管我们没有同质的 Pt-CO 类似物可以直接比较来证明这一点。有趣的是,从图2 B中可以看出,当催化剂在较低 pH 值下合成时,CO 谱带变宽(例如,合成 pH 值为 8 时半峰宽为 22 cm–1)并失去对称性,表明 Pt 的存在TiO 2载体上多个吸附位点上的异构体。似乎需要仔细控制合成条件才能将 Pt同种物质沉积在载体上的一致位点上。
虽然我们无法直接评论主要吸附 Ptiso的 TiO2位点的结构,但我们可以使用观察到的 CO 吸附强度来推断该位点的一些特征。CO 在 Ptiso/TiO2上的吸附能非常弱,可以从观察到的 CO 在室温惰性气氛中的容易解吸中推断出来。这与预测的以正方形平面结构吸附在CeO 2表面上的Pt异种对CO的弱吸附类似,其中计算出Ptiso被CeO2强烈吸附。(因此,假设本文报道的Pt异构体以非常强的吸附能结合到TiO 2载体上的一个位置(或几个类似位置),使得它们与载体的强配位降低了CO的结合能。这里报道的 Ptiso/TiO2的弱 Pt-CO 相互作用表明 CO 不会像之前观察到的那样使 Pt 原子移动,这与 CO 拉伸频率的严格 fwhm 一致,表明 Pt 主要位于载体上的单个位置。
之前的一份报告检查了 Ptiso物质在金红石 TiO2上的吸附位置,这表明 O 空位是最稳定的位点,尽管该信息不能与我们的工作直接相关,因为 TiO2载体纳米颗粒是 100% 锐钛矿。然而,考虑到在本研究中用作载体的 5 nm 直径锐钛矿晶体上预期存在小尺寸和高密度的缺陷,如果 Pt 同种物质的主要吸附位点是缺陷或台阶位点,也就不足为奇了。二氧化钛。应该指出的是,TiO2中的杂质可能为 Pt同种物质提供锚定位点,尽管此处使用的 TiO2中任何单一杂质的数量小于 Pt 的量,表明杂质没有提供稳定的结合位点。我们的结果表明,此处分析的 Ptiso物质本质上是单点材料,并且 Ptiso被 TiO2载体吸附得足够强,以最大限度地减少金属原子迁移率,从而与 CO 产生弱相互作用。
我们的结果出现了一个有趣的明显悖论,即 Ptiso物种和 Pt ox团簇上 CO 吸附能的显着差异。Pt ox簇由预氧化的 Pt金属簇的部分 CO 还原形成,并含有 Pt2+和 Pt0特征的 CO 吸附位点(图4和5)。我们认为,在这些部分氧化的 Pt 簇上,一些 Pt2+物种仍处于欠协调的几何形状,可能局部仅与 2 个氧原子结合,从而能够与 CO 强结合。相反,Ptiso物种与 CO 之间的弱结合表明这些Pt2+物质与TiO2中的氧原子强烈局部配位。我们认为,具有明显相似氧化态的两种 Pt 物质(可能是 Pt2+)由于其局部配位的差异而在与 CO 的相互作用强度上表现出很大的差异。这些细节将在未来进行更多探讨。
接下来,我们在之前关于其他载体上的 Ptiso 的报道的背景下,讨论 Ptiso /TiO2的稳态 CO 氧化反应性,以及 TiO2上 1 nm Pt金属簇的比较反应性。之前关于 Pt/FeO x催化剂上 CO 氧化的报告显示,与1 nm Pt 簇上的 Pt金属位点相比,Pt同位点的 CO 氧化 TOF 增加了约 2 倍,这与我们对类似 Pt 物种的结果非常一致在TiO2上。然而,与我们报道的 Ptiso /TiO2催化剂相比,Pt iso /FeO x 催化剂表现出明显更高的 CO 氧化 TOF ,其中在200 °C 下在 Ptiso/TiO2上观察到相同的 TOF (∼0.1 s –1 )在室温下观察 Ptiso /FeOx。最近,据报道,在校正 O2分压差异时, Fe2O3负载的 Ptiso表现出的 CO 氧化 TOF 比报道的 Ptiso/TiO2高约 4 倍。同一报告中还对 ZnO 载体的 Ptiso进行了检查,显示出与此处观察到的 TiO2几乎相同的 TOF(当校正 O2分压差异时) 。我们将这些可还原载体上 Ptiso的反应性差异归因于从载体中提取氧的活化能的差异,如下所述。
与可还原载体上的 Pt iso相比,Pt iso /TiO 2表现出比 Pt iso /θ-Al2O3显着更高的 CO 氧化反应活性和Ptiso/γ-Al2O3。尽管 Pt 实验中的氧分压高出 3.7 倍,但在 200 °C 下,Pt iso /TiO2的 TOF 比 Ptiso/θ-Al2O3高约 10 倍(0.013 s–1与 0.11 s–1 ) ,根据我们推导出的速率定律,表明 Pt iso /TiO 2上的 CO 氧化的固有 TOF 高 20 倍。类似地,尽管氧分压高4 倍,但与 Ptiso /γ-Al2O3相比,观察到 TOF 高约 4 倍,这表明 Ptiso/TiO 2上 CO 氧化的固有 TOF 大约高 8 倍。Ptiso /θ-Al2O3上的CO氧化和θ-Al2O3上的1 nm Pt金属簇的比较显示Pt金属簇的反应性高5倍。这可以通过 Pt iso催化循环中所需的 θ-Al2O3氧活化来理解,与 Al2O3负载的 Pt 簇上已知的 CO 氧化机制相比,这在能量上是困难的,其中整个反应循环发生在 Pt 上。
据报道,KLTL 沸石中的Ptiso物质在四胺 Pt 络合物氧化之前达到 0.0038 s–1的 TOF,在 150 °C、1% CO、5% O2中操作时,在催化剂氧化之后达到 0.012 s–1的 TOF ,和大气压下 94% He。在 Ptiso/TiO2上,在 150 °C 和 5 倍低的 O2分压下,测得TOF 为 0.015 s–1 ,这表明在相同的 O2分压下,Ptiso/TiO2催化剂的 TOF 约为 3 倍Ptiso/KLTL 催化剂活性更高。在这种情况下,没有与 Pt 团簇的反应性进行直接比较。我们发现,Ptiso的反应性高度依赖于载体,并且只有对于可还原氧化物负载的 Ptiso物种,Pt iso的 CO 氧化反应性才比 Pt金属簇增强。
为了解释文献中明显的 Ptiso的支撑依赖性反应性以及 Pt iso与我们观察到的 1 nm Ptmetal物种的位点依赖性反应性,我们考虑了观察到的 CO 氧化速率定律和活化势垒。TiO2上的Pt iso和1 nm Ptmetal金属催化剂都表现出相同的速率定律,其中O2分压呈半级依赖性,而CO分压呈零级依赖性。这与在类似条件下对 CeO2负载的 Rh、Pt、Pd 和 Ni 颗粒进行的 CO 氧化测量一致。我们通过原位红外观察到,Pt同种物质对 CO 的稳态覆盖率较低,而 Pt金属位点在反应条件下对 CO 的覆盖率很高。然而,在反应条件下观察到CO在Pt金属位点上的覆盖并不意味着CO在Pt金属簇和TiO2表面之间的界面处有显着的覆盖,其中Pt和TiO2上的氧直接键合使 Pt 原子呈阳离子。
这些结果表明,反应通过 TiO 2负载的 Pt 结构上的 Mars van Krevlen 机制进行,其中限速步骤涉及原子氧从 TiO 2迁移到 Pt iso或 Pt金属簇上的界面 Pt 原子上,即称为反向氧气溢出。O2分压的半阶反应依赖性表明O2解离不太可能是速率限制步骤,因为这将产生一阶O2依赖性。催化循环中发生的从TiO2到界面Pt原子的反向氧溢出并且是限速步骤的假设与后过渡金属促进氧化物还原性的众所周知的行为一致。例如,从微动力学模型推断,在Pt/TiO2催化剂上的水煤气变换反应条件下,Pt/TiO2界面处不可忽略的氧空位浓度可以是稳定的。这与相同条件下裸露的TiO2上的低空位浓度形成对比。使用氧同位素标记的实验研究也表明TiO2中的氧原子作为Pt/TiO2催化剂上的水煤气变换反应中的活性物质。
除了Pt可以促进TiO2的还原性的证据之外,还有强有力的证据表明相对容易的反向氧溢出从氧化物到小金属簇和单个原子。例如,通过实验和理论计算已经证明,将氧溢出从CeO2逆转到Pt(或Rh)是一个放热过程,并且可以在温和条件下发生。此外,通过理论计算表明,对于 TiO2上的小 Ru 和 Ni 团簇,反向氧溢出在能量上是热中性或放热的,并且进一步假设该过程在 TiO2纳米粒子上将更加放热,如在我们的实验与模型中使用的扩展 TiO 2表面进行了比较。因此,我们得出结论,氧从 TiO2迁移到界面 Pt 物质在能量上是可行的,并且是在探索的 CO 氧化条件下的限速步骤,与我们的分压相关测量一致,并且与之前反向氧溢出分析和金属促进氧化物还原性的已知能力。
除了解决速率对 O2分压的半阶依赖性之外,重要的是从机理上理解如何不依赖于 CO 分压。缺乏CO分压依赖性要求气相CO和吸附CO都不参与限速步骤,并且CO在活性位点上的覆盖率基本上为零。对于 Pt同种活性位点的情况,原位红外分析显示 Pt同种物质上没有可测量的 CO 覆盖,这与基于观察到的分压依赖性的动力学评估一致。然而,在 1 nm Pt金属簇上,在反应条件下观察到显着的 CO 覆盖。据认为,由于与TiO2表面的氧原子配位而呈阳离子的界面Pt原子与存在于远离界面的簇上的金属Pt原子相比表现出降低的CO结合能。界面 Pt 原子处的 CO 结合能降低,与 Pt 簇和载体协调,最大限度地减少了反应条件下的 CO 覆盖范围,从而实现了与 Pt 同种物种观察一致的机制和速率定律。
载体氧参与 Ptiso物种的 CO 氧化催化循环表明,在 Ptiso上测量的 CO 氧化的依赖于载体的 TOF (通过与文献的比较推断)可能源于与从载体中提取原子氧相关的不同活化能。通过考虑测量的 CO 氧化E app以及与之前测量结果相比的速率定律,可以进一步理解这一点。在与本文报道的类似条件下,负载在 Al2O3上的 Pt金属簇对 CO 的氧化显示出约 85 kJ/mol 的E app,速率定律在 O 2中为一阶,在 CO 中为负一阶,并且进一步显示显着TOF 低于此处报道的 Pt/TiO2。这些差异证明整个反应循环发生在 Pt金属簇上的 WC 位点上,并且 Pt金属表面被 CO 饱和。与 Pt iso和 Pt金属位点的 Pt/TiO2结果比较(其中观察到不同的速率定律和降低的E app)强烈表明,在相同条件下,可还原和不可还原氧化物负载的 Pt 结构的 CO 氧化机制是不同的,差异源于载体参与可还原氧化物的催化循环。假设与TiO2上的 Pt iso相比,观察到的 1 nm Pt金属簇的E app较低(53 vs 68 kJ/mol),这是由于 Ptiso物种对 TiO2还原性的影响较弱,因此氧迁移到 Pt 位点的屏障增加。即使 Pt iso上的势垒增加,CO 氧化的 TOF也大于 1 nm Pt金属簇,这表明与1 nm Pt金属簇。
基于上述分析,我们提出,1 nm Ptmetal簇和 Ptiso物质的催化 CO 氧化循环涉及限速步骤中从 TiO2到界面 Pt 原子的反向氧溢出。这些结构中的界面Pt原子被认为与其局部环境和阳离子良好协调,导致CO吸附能相对减弱,从而减轻了相似条件下金属Pt原子众所周知的CO中毒。需要指出的是,如果阳离子 Pt 原子存在于配位不足的局部环境中,从而与 CO 结合得更牢固,如我们的 Ptox案例所示,这些位点将被 CO 毒害,直到氧气被夺取形成金属位点。 。因此,Pt 原子上的电荷与局部环境的配位之间的仔细平衡,除了存在于与可还原氧化物的界面处以实现反向氧溢出之外,对于在可还原氧化物负载的 Pt 结构上形成最活跃的位点是必要的。
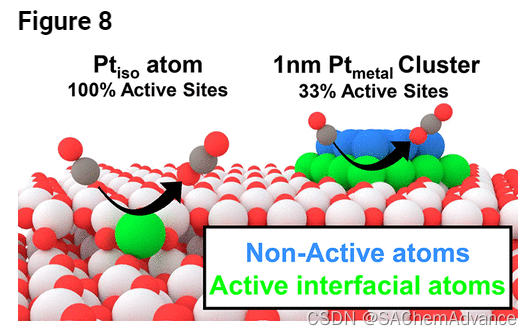
这一假设与最近分析 CeO2上 2 至 20 nm Pt、Pd 和 Ni 颗粒的工作一致,其中开发了一个模型,将界面金属原子的浓度与 CO 氧化 TOF 相关联,但将这种趋势扩展到极限单个金属原子的活性位点。当将团簇从 2 nm 缩小到单个原子时,界面处存在的总金属原子的比例增加了 10 倍,这显着提高了金属利用率。Ptiso上每克 Pt 的 CO 氧化速率和 TOF与 TiO2上的1 nm Pt金属簇的比较支持了这一结论,其中所提出的 1 nm Pt 簇结构如图8所示。假设界面 Pt 原子是唯一的活性位点,与 1 nm Pt金属簇相比,仅含有 Pt 同种物质的催化剂预计每克 Pt 的活性高 3 倍,而测量到的差异为 4-6 倍;类似地,预计Pt iso的 TOF 比 1 nm Ptmetal簇高 1.75 倍,而测量到的差异约为 1.12–2 倍。考虑到计算的速率和 TOF 假定 1 nm Pt 簇结构,而 STEM 分析证实 1 wt% DI 催化剂中存在一些较大的 Pt 颗粒,并且存在两个位点(Ptiso与 Ptmetal)的活化势垒存在微小差异,这是由于它们诱导载体还原的能力不同。因此,得出的结论是,当负载在 TiO2上时,Ptiso /TiO2催化剂是 Pt 在 CO 氧化中最有效的利用,因为每个原子都暴露于催化作用并与载体界面,通过反向为催化循环提供相对移动的 O 原子。氧气溢出并削弱CO结合能。
5.结论
总之,我们展示了一种合成方法和催化剂结构,其中 Pt同种物质以每个 TiO2颗粒 <1 个 Pt 原子的比例分散在直径 5 nm 的TiO2颗粒上,从而产生在各种条件下稳定的 Pt同种物质。使用相关的 STEM 成像和 CO 探针分子 IR 表征方法,识别了与 Ptiso、 Ptox簇和TiO2上的 Ptmetal簇结合的 CO 的红外特征,从而可以快速识别和表征每种类型的位点。结果发现,通过 SEA 合成方法产生的 Pt同种物质基本上是单位点物质,这是从吸附 CO 伸缩带的紧密〜6–8 cm –1 fwhm 推断出来的。使用推断的光谱特征,这些位点上 CO 吸附能的顺序被确定为 Ptiso ≪ Ptmetal< Pt ox,其中 Pt ox上吸附的 CO在低于约200 °C 时基本上不具有 CO 氧化活性。对 Ptiso和 1 nm Ptmteal簇上稳态 CO 氧化的严格动力学测量表明,Ptiso位点本质上更具反应性,因为催化剂上的每个 Pt 原子都与发生反应的载体接触并暴露于反应物。这项工作表明,某些可还原载体上的 Ptiso为 CO 氧化提供了最有效的金属利用,并且进一步表明,本文提出的结构和合成方法通常可用于探测分离的先前金属原子催化剂的光谱特征和反应性。
论文地址:Catalyst Architecture for Stable Single Atom Dispersion Enables Site-Specific Spectroscopic and Reactivity Measurements of CO Adsorbed to Pt Atoms, Oxidized Pt Clusters, and Metallic Pt Clusters on TiO2