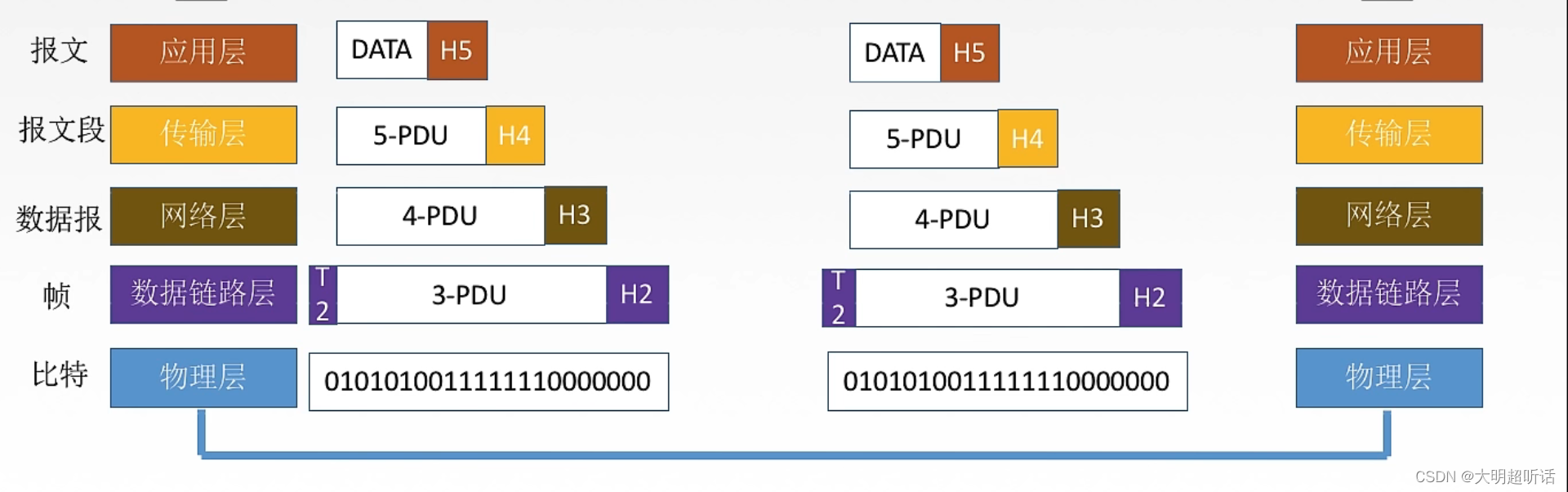
生物信息学
生物信息学——基础篇——一至三代测序技术
文章目录
- 生物信息学
- 一、一代测序
- 二、二代测序
- 三、三代测序
- 四、总结
一、一代测序
-
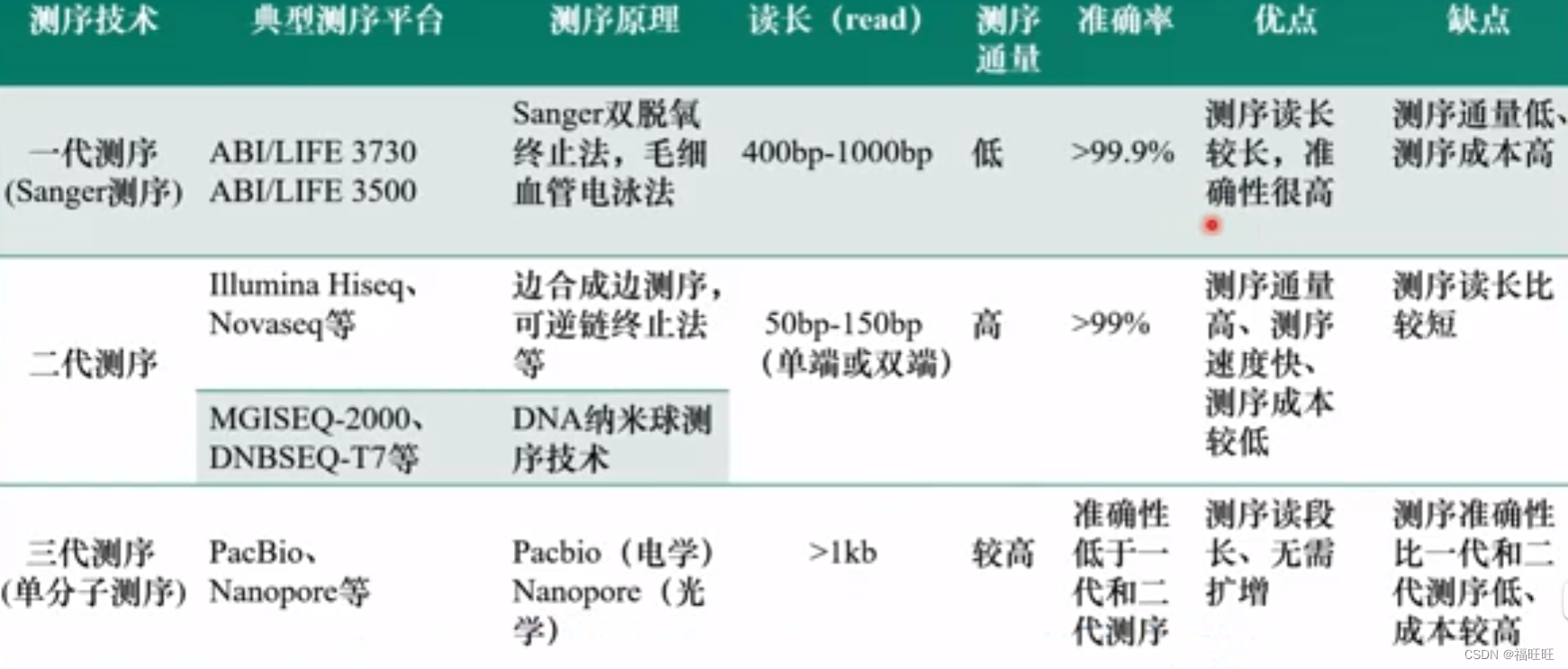
概述:一代测序(又称Sanger测序)。
-
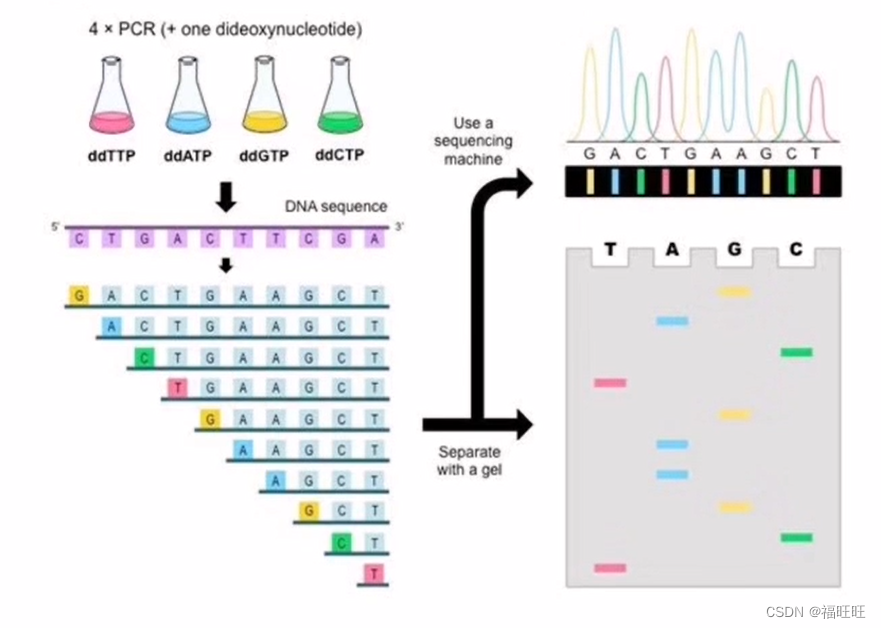
原理:Sanger测序利用一类特殊的核昔酸,即ddNTP (双脱氧核苷三磷酸):这种核昔酸不能形成磷酸二酯键,故会中断DNA的延长。如果用同位素对ATCG四个碱基进行部分标记,电泳后通过放射显影就可以知道这4个碱基出现的位置。例如针对序列ATCGATCGATCG,对A作为终止标记。即可得到三类片段,即A,ATCGA,ATCGATCGA。因此分析可以知道碱基A出现在第1位,第5位和第9位。同样,如果用同样的方法对其他碱基进行标记,也可以测定其余碱基的位置,最终获得待测序列的碱基顺序。
-
过程:
-
优点:操作简单,误差小。
-
缺陷:成本高,通量低,一次只能测一条DNA,而且测序长度有限(至多约为1000bp)。
二、二代测序
- 概述:二代测序(又名高通量测序),主要是为了解决一代测序长度限制,且通量低效率低的问题而开发。目前,二代测序(主要为Illumina测序仪)是市场主流。
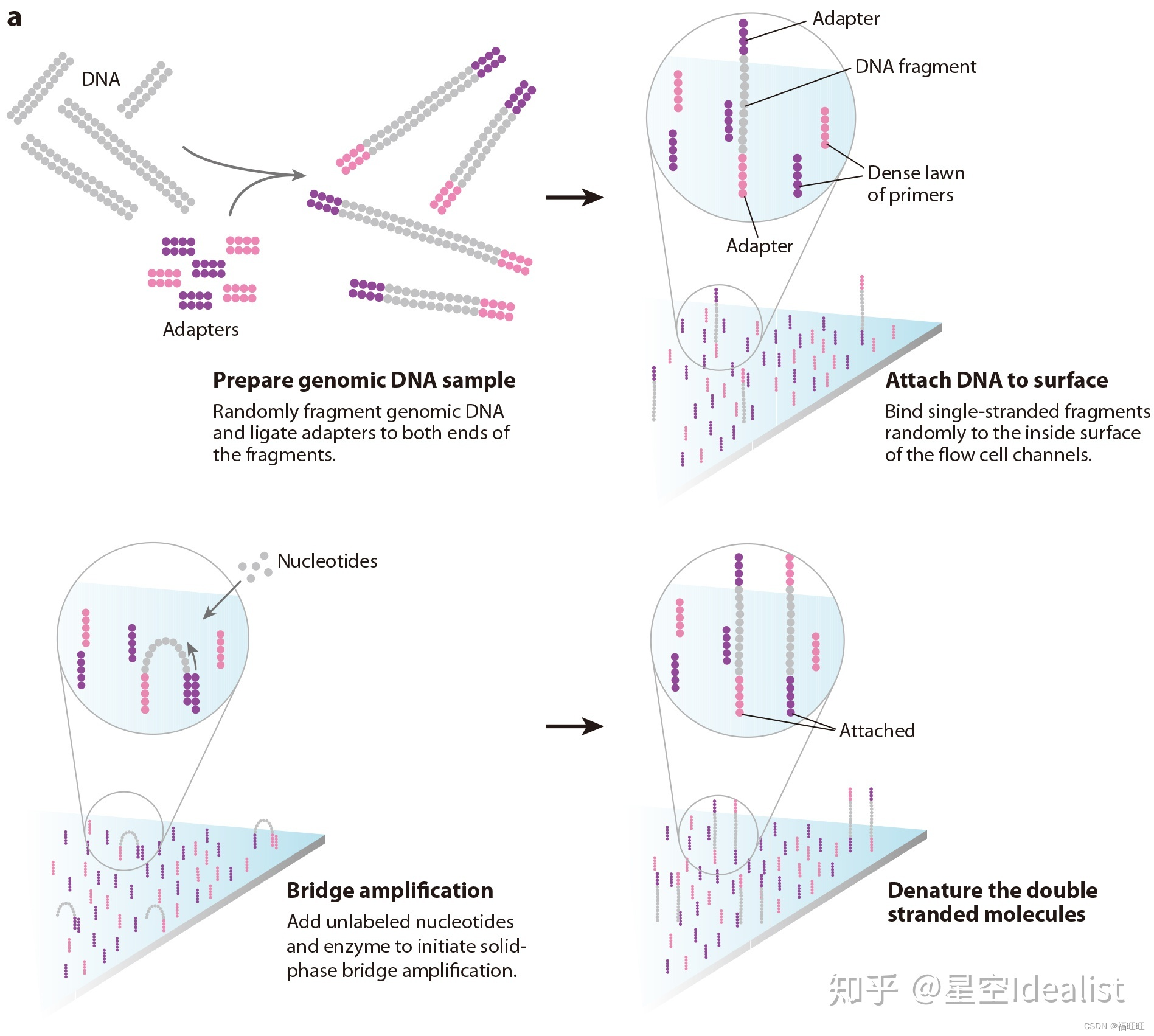
- 原理:将长的DNA(或者RNA反转录的cDNA)进行碎片化(鸟枪法),之后利用接头将片段化的DNA固定在不同材质的表面。之后进行桥式PCR形成簇状结构,该过程扩增了DNA,相当于放大了信号强度。之后使用不同荧光基团对4种碱基进行标记,就可以根据荧光信号确认新合成的DNA单链时连接上去的碱基。
- 过程:
- 文库构建:即为测序片段添加接头。无论是PCR产生的片段还是基因组鸟枪法打断的片段都具有特异性(PCR中不同样品反向引物插入了特异性的barcode,因此两端也是特异的),两端缺乏必要的引物因此混合DNA片段不能直接扩增和测序。DNA片段需要加接头修饰才能进行上机测序,这个过程称为二代测序的文库构建。
①末端修饰。目前很多PCR使用的高保真Pfu聚合酶产生的片段末端是平齐的(也即没有不配对的碱基);鸟枪法产生的片段则是随机断裂,其末端可能是平齐的也可能是不平的。因此,建库第一步是使用Taq聚合酶补齐不平的末端,并在两个末端添加突出的碱基A,从而产生粘性末端(若使用Taq酶扩增,则无需末端修饰),产生粘性末端的片段可以添加接头(Adaptor)。
②添加接头。经过末端修饰后的PCR片段末端具有突出的A尾,而接头具有突出的T尾,可以使用连接酶将接头添加到DNA片段两端。NEB的接头为特殊的碱基U连接的环状结构(可以增强稳定性),因此连接接头后,还需要将碱基U删除从而形成“Y”形接头。这一步添加的接头主要是为了后续PCR中作为引物扩增继续添加文库index和与测序平台互补的寡核苷酸序列(此外还作为测序引物Rd1 SP/Rd2 SP),而之所以为“Y”型开叉结构,是因为每一端接头是两条不互补的序列(每一端都是Rd1 SP与Rd2 SP交错),因为连接酶没有选择性,每个接头都是只靠突出的T来与DNA连接,“Y”接头保证了每条单序列两端均为不同的测序引物,从而在后续PCR中可以连接不同的寡核苷酸序列(P5/P7),(每个DNA短片段的正链与反链都加上了P5与P7,因此建库后每个DNA片段都会扩增出两种结果)具体流程见下图。
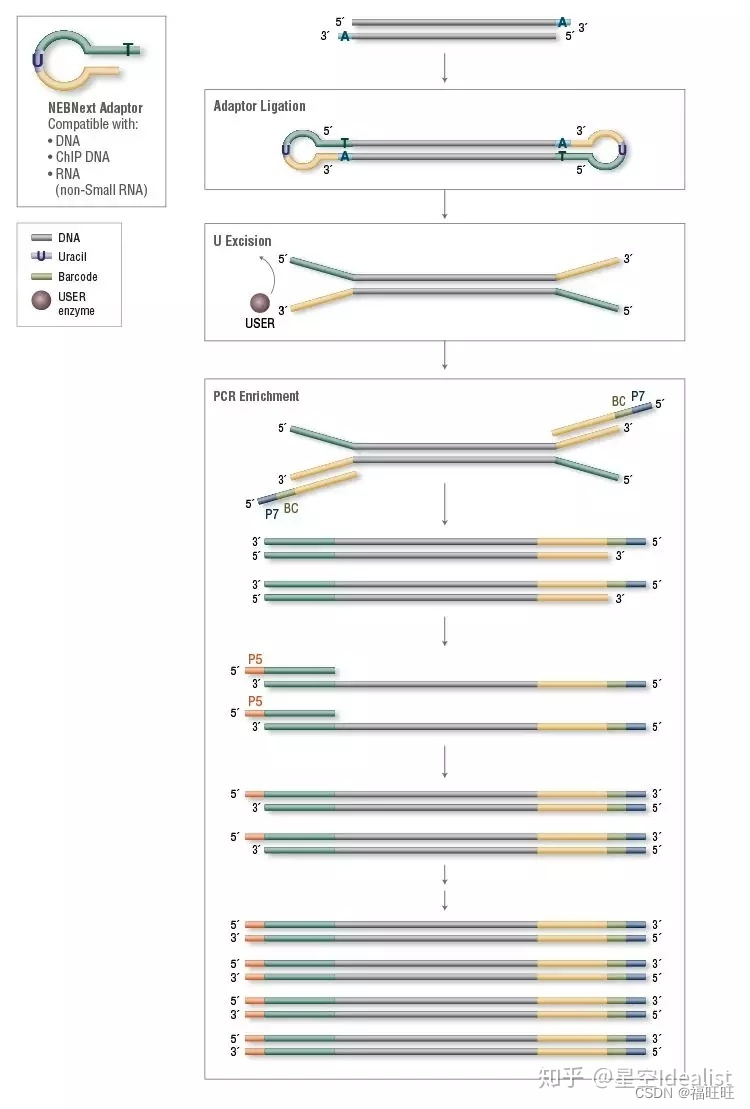
③磁珠纯化。添加接头后的文库体系中含有聚合酶、连接酶等各种酶以及辅助物质,接头的添加也是过量的,而且由于末端的不稳定性,容易形成自连片段,鸟枪法打断的片段中也可能有大片段存在,所以需要特殊磁珠(AMPure XP Beads)纯化来去除大片段以及各种杂质,从而获得成功添加接头的文库片段。其原理为磁珠可以通过氢键等作用力来吸附DNA片段,磁珠本身不具有片段大小选择的能力,但其储存的buffer里面含有20%的PEG 8000,PEG浓度越大则可以吸附的DNA片段越小。因此磁珠纯化的时候要根据文库片段不同严格控制磁珠添加量(其实是PEG添加量)来实现片段选择。
④PCR扩增。添加了接头的DNA片段,可以使用与接头互补的引物来扩增。这个过程非常重要,因为目前所有片段其两端是不互补的Y形结构,不能直接进行测序;此外,片段还需要添加用于区分不同文库的特异性index,以及与测序仪芯片互补的两种寡核苷酸序列(P5/P7)。
⑤第二次磁珠纯化。PCR后需要将产物DNA片段与聚合酶等杂质分离,因此再次进行磁珠纯化,之后进行质量检测,包括DNA浓度检测、琼脂糖凝胶电泳和片段长度检测,完成建库。测序是以单链为单位的,建库完成后的每条DNA的单链均一端连有测序引物Read1 Sequencing Primer(Rd1SP)和P5,另一端为Rd2 SP、Index(Barcode)和P7。Index用来区分不同的文库,因为测序仪一个run产生数据量巨大,由于实际情况不同,一次上机常会进行多个文库测序,因此需要加上Index来区分。
- 上机测序:
Illumina测序技术为基于基因芯片的边合成边测序,整个平台可解剖为三个系统:一温度控制系统,原理和普通PCR仪一样,来控制反应的进行;二酶控制系统,通过各种酶来控制DNA合成与剪切;三荧光信号收集系统,可以理解为分辨率极高的照相机。在Illumina测序平台的流通池(Flow cell)表面,通过基因芯片技术交错固定了无数条寡核苷酸链(即短核苷酸链),分别为P5’(P5互补)和P7,单链化的文库DNA片段进入流通池后,包含P5或P7’的单链可以与表面的寡核苷酸基于互补配对结合,从而进入测序过程。测序具体流程如下:
①首先以寡核苷酸为引物、文库片段为模板进行DNA复制(因为文库稀释后浓度足够低,可以认为文库片段均匀的结合在流通池表面,每个片段结合的位置相距足够远,这很重要,否则测序时会导致信号叠加而不能识别)。复制完成后解链,将文库片段洗去,留在流通池表面的为与文库模板互补的DNA链。
②因为单链DNA另一端为不同的接头序列,可以与相邻的另一种寡核苷酸互补结合,之后进行“桥”式扩增(假如第一次结合的为P7,则复制完成洗脱模板后顶端可以与相邻的P5互补结合形成“桥”,并以P5为引物进行复制,完成后再次解链并与相邻不同种接头结合来进行复制,如此类推)。25-28个循环完成后,原来散布在表面的单核苷酸序列变成散布的DNA簇,这一步主要是为后续测序做准备,因为测序时单分子产生的光信号很弱,难以检测。
③“桥”式扩增后一个DNA簇都是由最初的一个文库模板复制而来,但是这时候P7上的序列与P5上的序列是分别从两端开始的,测序要保证每个片段一致性(都是正向或都是反向),因此再次解链线性化,切割并洗去P5上的DNA链,只留P7上的DNA单链。Illumina巧妙地利用了甲酰胺基嘧啶糖苷酶Fpg对8-氧鸟嘌呤糖苷8-oxo-G的选择性切断作用,在合成的引物链上加入了一个8-oxo-G,用Fpg处理,就把带8-oxo-G基团切掉,并把DNA链切断,留下一带不完整糖基的磷酸基。这个磷酸基在接下来的过程中,起到了阻止P5延伸的作用。此后的双末端测序中需要恢复3’-OH,则用脱嘌呤嘧啶内切核酸酶AP-endonuclease把带不完整糖基的那个磷酸基切掉。
④加入测序引物Read1 SP和修饰过的DNA聚合酶,则在测序引物3’端开始DNA复制。在流通池加入可逆终止荧光dNTP,其3’-OH被阻隔(糖基3’连接有叠氮基团,在链延伸时起到了阻止添加下一个dNTP作用,因此在除去阻隔前只能添加一个碱基),4种dNTP在碱基上分别连接有不同颜色的荧光基团(也可以相同颜色荧光标记,但是测序会更慢,每次只能添加一种碱基)。之后洗掉多余的dNTP,使用激光扫描,收集留在流通池表面的荧光信号(如图1-6所示)。用巯基试剂去掉3’位阻断的叠氮基团,用TCEP(Tris(2-carboxyethyl)phosphine,三(2-羧乙基)膦)去掉荧光基团,进入下一个碱基的测序反应。因为每条DNA单链扩增形成的DNA簇均固定在表面,随着反应进行根据相同位置出现的荧光信号情况,就逐渐读出了改位点DNA链的序列。
⑤要保证测序的准确性,需要一个位点DNA簇的每条链同步复制,然而随着反应进行,不同链复制情况会出现差异,因此二代测序读长目前限制在300bp以内。Read1结束后,解链并洗掉测序中已经合成的部分,加入测序引物Index引物(也即Read2 SP互补的寡核苷酸),这时会继续在3’端进行复制,读出接头中Index序列,从而可以确定出每个位点的DNA属于哪个文库。
⑥为了增长测序长度,进行另一个方向测序,也即双末端测序。洗掉前面复制合成的片段,DNA单链继续在流通池表面形成桥式连接,这时要用脱嘌呤嘧啶内切核酸酶处理修复P5的3’-OH末端,加入聚合酶,则在P5末端开始DNA复制。十几个循环后,将P7上的DNA切割并洗掉。Illumina通过在P7核酸链中加入一个U碱基,用USER酶(Uracil Specific Excision Reagent,尿嘧啶链特定切断试剂)来切隔断链。这时只留下P5上的DNA链,与Read中方向相反。加入测序引物Read2 SP,进行另一端的序列读取。
- 测序数据:得到测序数据为fastq格式的碱基序列
- 优点:
- 与第一代测序合成完进行电泳,显影最后得到结果相比,边合成边测序的测序效率得到极大提高。
- 将长DNA片段化后,可以同时进行多个通道的测序,提高通量,减少时间。
- 缺陷:
- 每一片段的读长较短(约为500bp)。
- 进行长DNA测序时,需要对结果进行拼接,同样在边合成边测序的策略下,误差来源就是合成时碱基突变(GC突变等)。这就导致对于高度杂合的基因组、高度重复序列、高GC的区域、拷贝数变异等测序,往往不能利用二代测序准确获得。
三、三代测序
- 概述:三代测序(又名单分子测序),即理论上可以进行超长读长,不需要进行PCR扩增的测序。以SMRT (Pacific Biosciences)技术和Oxford Nanopore为代表。
- 原理:SMRT技术采用边合成边测序的思路,以及荧光检测的基本技术。
- 过程:SMRT中,碱基采用4种可断裂的荧光标记基团,当被DNA聚合酶合成到新生成的DNA链上时,荧光基团发生脱落,检测不同荧光脱落信号,即可获得序列。然而这一操作很容易被背景杂光干扰,因此该技术中还有一个重要组成部分就是零模波导孔,这是一个足够小的小孔(10nm) ,小到只能插入一个DNA聚合酶和一条DNA序列,因此在检测的时候可以不受到背景干扰,只探测到一条DNA的信号。
- 优点:
- 超长读长(10kbp)
- 样品用量极少
- 适用于长片段和高度多样性的基因组
- 测序速度快
- 缺陷:
- 需要构建文库
- 误差率比较高,需要多次测序来分析和降低误差。
- 原理:Oxford Nanopore技术即纳米孔技术,这一技术直接绕开了边合成边测序和荧光检测这两个传统思路。
- 过程:四种碱基由于有不同的结构,因此带电特点不同,通过纳米孔时引起的电流变化不同。通过测量电流变化来观测到不同分子穿过纳米孔。而通过分析ATCG四种碱基的特点,即可绘制测序结果图。其本质是检测变化的电信号。纳米孔的成分可以是生物纳米孔,即使用蛋白质镶嵌在膜上(Oxford Nanopore Technologies技术使用溶血素蛋白),也可以是化学纳米孔,如石墨烯,高分子材料孔。
- 优点:
- 理论上无读长限制
- 不需要额外碱基,可以直接测定RNA,再也不用逆转录(都是检测电信号)
- 样品用量极少
- 缺陷:
- 误差比较大(长DNA可能在孔口出现缠绕,且过孔控制不好)
四、总结
- 一代技术,误差低,读长中等,但是速度慢,效率低。
- 二代技术读长短,但是可以同步进行多个片段测序,效率非常高。(目前主流测序技术)
- 三代测序读长超长,效率更高,且需要样品量极少(单分子),但在目前发展阶段,误差比较高。