命令解释参考GROMACS基本教程
整个流程参考分子动力学模拟Gromacs一般使用步骤(空蛋白)
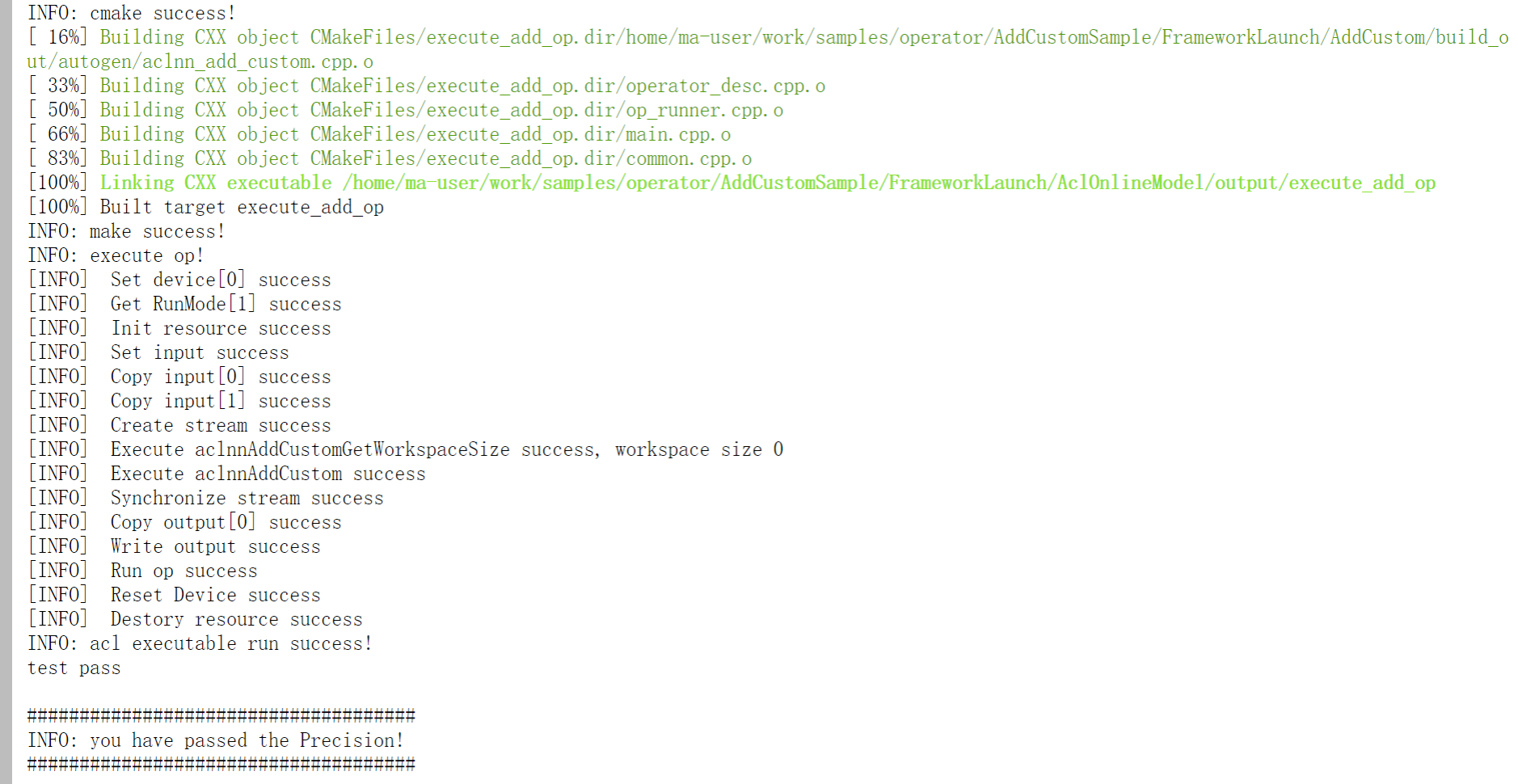
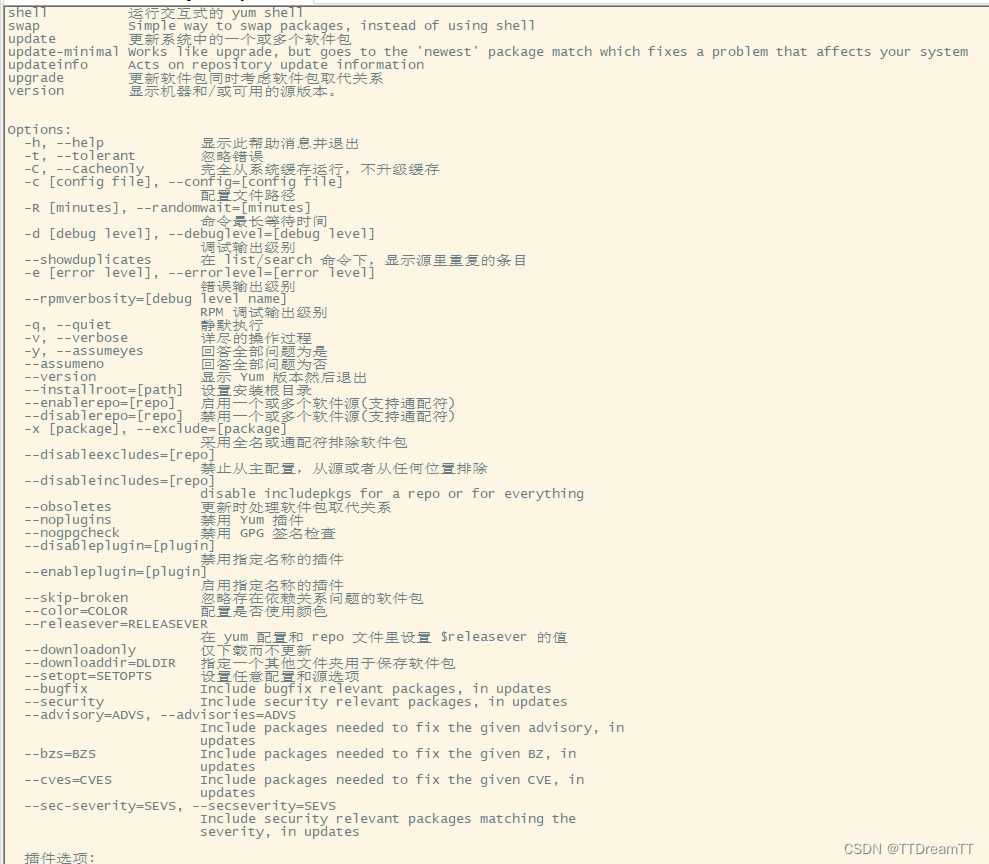
从gromacs 5.0版本开始,所有的工具都是“gmx”的子模块。可以通过下面的命令获得任何一个模块的帮助信息:
gmx help (module) 或者 gmx (module) -h
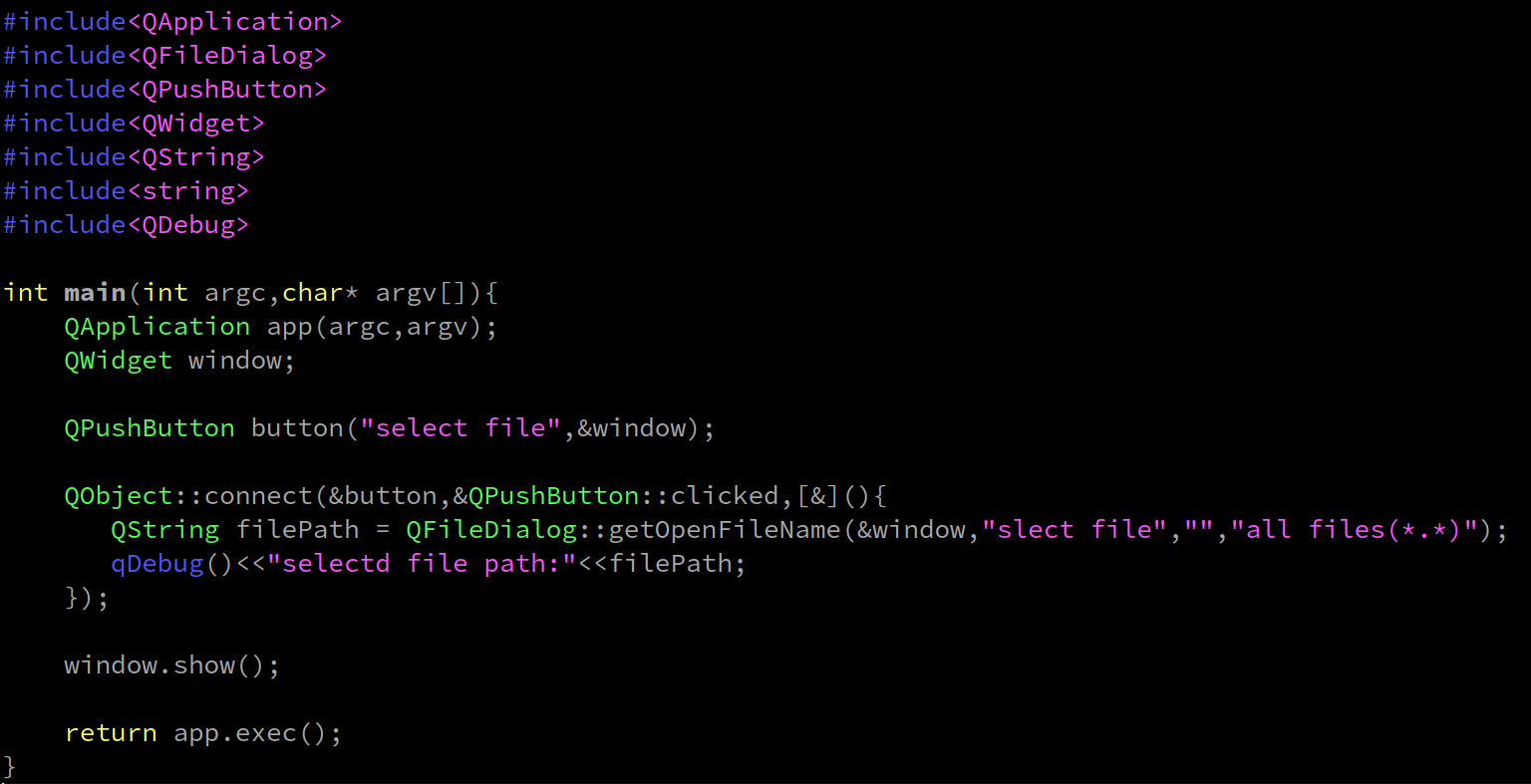
#!/bin/bash
module load gromacs/gromacs-2021.5-gcc-9.2.0-cuda11.1
for i in *.pdb;do
pdbid=${i:0:7}
echo $pdbid
printf "8"|gmx pdb2gmx -f $i -o ${pdbid}_processed.gro -water spc -ignh
gmx editconf -f ${pdbid}_processed.gro -o ${pdbid}_newbox.gro -bt dodecahedron -d 1.0
gmx solvate -cp ${pdbid}_newbox.gro -cs spc216.gro -p topol.top -o ${pdbid}_solvate.gro
gmx grompp -f em.mdp -c ${pdbid}_solvate.gro -p topol.top -o next.tpr -maxwarn 1
printf "13" | gmx genion -s next.tpr -o ${pdbid}_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
gmx grompp -f em.mdp -c ${pdbid}_solv_ions.gro -p topol.top -o em.tpr
gmx mdrun -v -deffnm em
gmx editconf -f em.gro -o em.pdb
done
1.用pdb2gmx 处理 pdb 文件
gmx pdb2gmx -f $i -o ${pdbid}_processed.gro -water spc -ignh
(1)pdb2gmx此命令将pdb文件转换成gromacs文件并产生拓扑文件。
(2)-ignh因为本pdb文件是由 NMR产生的,含有氢原子,因此用-ignh选项忽略文件中的氢原子。
(2)注意-ignh选项, 它表明程序会首先移除初始文件中的氢原子, 然后再根据相应力场的说明进行重建。???
(3)-f读入pdb文件
(4)-ff可指定力场(G43a1是Gromos96力场,一个通用原子力场)(没用过)
(5)-o指定一个新产生的pdb文件(也可以是其它多种类型文件)的文件名
(6)-p指定新产生的拓扑文件名。拓扑文件包含了所有力场参数(基于一开始选择的力场),因此非常重要(也没用过)
(7)-water来指定水模型研究表明SPC/E 水模型在水盒子模拟中表现最好。用SPC/E 水模型研究长程静电相互作用较好。
(8)注:对于下面将要用到的任何命令,都可以使用“-h”查看该命令的使用方法,比如,对于命令pdb2gmx 可以使用: pdb2gmx –h
补充: pdb2gmx的目的是生成以下三个文件:
(1)分子的拓扑文件;(2)位置限定文件;(3)预处理的结构文件。
(4)拓扑文件(默认为topol.top)包含了模拟中定义一个分子的所有必须信息,包括非键相互作用(nonbonded,原子类型和电荷)和成键参数(bonded, 键、角、二面角)。后面生成相应文件后再详细查看。
(5)-ter:对N-端和C-端的电荷进行交互分配
(5)使用-ter选项后GROMACS会提示分子的末端变化, 合适的选择应该能够正确地表示体系。???
(6)-inter:对Glu、Asp、Lys、Arg和His的电荷进行交互分配;选择那(哪)些Cys参与二硫键
(7)xxx_processsed.gro是GROMACS格式的结构文件,包含了力场文件中包含的所有原子(蛋白中氨基酸的H原子已经补全)。
(8)topol.top文件是系统拓扑文件(接下来会详细介绍)。
(9)posre.itp包含了限制重原子位置的信息(后面会详细介绍)蛋白的位置限制文件posre.itp
(10)最后要注意一点:很多用户认为.gro文件是必须的。其实不然。GROMACS可以处理很多格式的文件,只不过.gro是默认的坐标文件格式。.gro文件非常紧凑,但精度不够高。如果你想用PDB等其他格式的文件,只需要指定.pdb后缀的输出文件名即可。pdb2gmx的目的是生成符合力场的拓扑文件;输出结构在很大程度上是此目的的副产品,目的是为了方便用户。具体格式几乎可以是任何你喜欢的格式(不同的格式可以在GROMACS手册中查看)。
2.建立盒子
gmx editconf -f ${pdbid}_processed.gro -o ${pdbid}_newbox.gro -bt dodecahedron -d 1.0
(1)-d决定了盒子的尺寸,即盒子边缘距离分子边缘 1.0nm (1.0Å)。理论上在绝大多数系统中,-d 都不能小于0.85nm。
(2)注:editconf 也可以用来进行gromacs文件(.gro)和pdb 文件(.pdb)的相互转化。
例如:gmx editconf –f file.gro –o file.pdb 则将file.gro 转换为 file.pdb
(3)现在就可以用产生的文件进行真空模拟了。真空模拟就是先能量最小化,然后进行动态模拟。
没用过下面图片中的命令
(4)-c会将蛋白质放在盒子的中心,而且蛋白质离盒子边缘至少1.0 nm(-d 1.0)。盒子的类型定义为立方型(-bt cubic)。距离盒子边缘的距离是一个非常重用的参数。由于需要使用周期性边界条件,我们必须满足最低镜像约定(minimum image convention)。也就是说,蛋白质不能与它的周期性镜像相遇,否则此时计算得到的力是不正确的。定义溶质-盒子的距离为1.0 nm就意味着蛋白质的两个周期性镜像之间至少有2.0 nm。这个距离可以满足模拟中几乎所有的截断方法(cutoff scheme)。
(5)看看editconf的输出, 注意体积的变化. 另外, 也看看protein-PBC.gro文件的最后一行(提示: 可使用命令tail -n 1 protein-PBC.gro). 在GROMACS格式(.gro)中, 最后一行指定单位晶胞的形状, 并总是使用三斜矩阵的表示方法, 其中前面的三个数字指定对角元素(xx, yy, zz), 后面的六个数字指定非对角元素(xy, xz, yx, yz, zx, zy).
gmx solvate -cp ${pdbid}_newbox.gro -cs spc216.gro -p topol.top -o ${pdbid}_solvate.gro
(1)通过solvate模块填充水分子(之间的版本是genbox)
(2)“我强烈建议使用十二面体(rhombic dodecahedron),因为它的体积大概是立方盒子的71%。因此,在溶剂蛋白质时可以减少大量水分子。”
(3)使用spc216.gro,这是一个通用的平衡过的3粒子溶剂模型
(4)spc216.gro可以适用于SPC、SPC/E或者TIP3P水模型,因为他们都是3粒子溶剂模型
(5)solvate模块可以追踪添加了多少水分子,然后写入到拓扑文件中以反映做出的改变。注意,如果使用的是其他非水溶剂,solvate不能自动更改拓扑文件!它与更新水分子的兼容性在代码中是固定写死的。

4.设置能量最小化
em.mdp文件:Gromacs用*.mdp 文件指定所有计算的参数。
title–标题随便取(最长64个字,简单点好) cpp–指定预处理器的位置define–传递给预处理器的一些定义。 –DFLEXIBLE告诉grompp将spc水模型而非刚性SPC包含进拓扑文件, 以便用最陡下降法进一步最小化能量。constraints–设置模型约束 integrator– steep,告诉gompp使用最速下降法进行能量最小化。 cg则代表使用共轭梯度法。dt–能量最小化用不用。只在动力学模拟中用(如md)。 nsteps–在能量最小化中,指定最大运行步数。 nstlist–更新邻居列表的频率。 nstlist = 10表示每10步更新一次。 rlist–短程邻居列表的阈值。 coulombtype–告诉gromacs如何计算静电。 PME为particle mesh ewald法(参见Gromacs用户手册) rcoulomb–指定库仑力阈值vdwtype–告诉Gromacs如何计算范德华作用(cut-off, Shift等) rvdw–指定LJ或Buckingham势能距离阈值 EM Stuff emtol–最大的力如果小于此值则能量最小化收敛(结束)(单位kJ mol–1 nm–1) emstep–初始步长(nm)
5.用grompp程序进行文件处理
gmx grompp -f em.mdp -c ${pdbid}_solvate.gro -p topol.top -o next.tpr -maxwarn 1
(1)-f标签指定输入参数文件(.mdp)
(2)-c输入结构文件(pdb文件,.pdb)
(3)-p输入拓扑文件
(4)-o输出mdrun的输入文件(*.tpr)
(5)topol.top拓扑文件内容:参考GROMACS教程1:Lysozyme
nr:原子序号type:残基序号 residue:残基名称(注意PDB文件中残基名称是“LYS”;.rtp文件中的“LYSH”表明残基被质子化) atom:原子名称 cgnr:电荷分组序号(charge group定义了整数电荷的单元,对加速计算有利) charge:原子电荷(“qtot”是分子上电荷的总和) mass:原子质量typeB、chargeB、massB:用于计算自由能微扰
; Include forcefield parameters
#include "charmm27.ff/forcefield.itp"
[ moleculetype ]
; Name nrexcl
Protein_chain_A 3
[ atoms ]
; nr type resnr residue atom cgnr charge mass typeB chargeB massB
; residue 1 GLY rtp GLY q +1.0
1 NH3 1 GLY N 1 -0.3 14.007
2 HC 1 GLY H1 2 0.33 1.008
3 HC 1 GLY H2 3 0.33 1.008
4 HC 1 GLY H3 4 0.33 1.008
5 CT2 1 GLY CA 5 0.13 12.011
6 HB 1 GLY HA1 6 0.09 1.008
7 HB 1 GLY HA2 7 0.09 1.008
8 C 1 GLY C 8 0.51 12.011
9 O 1 GLY O 9 -0.51 15.999 ; qtot
(6)grompp的作用是运行坐标文件和拓扑文件,生成原子级别的输入文件(.tpr)。.tpr文件包含了体系中所有原子的参数。
(7)为了通过grompp生成.tpr文件,这里需要一个额外的.mdp输入文件(molecular dynamics parameter,检查mdp);grompp会收集.mdp文件中的坐标和拓扑信息来生成.tpr文件。
6.使用genion和tpr文件添加离子
gmx genion -s next.tpr -o ${pdbid}_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
(1)对生成的tpr文件加入补偿离子以中和系统中的净电荷。
(2)-nname指定阴离子名称(在Gromos G43a1力场中,用CL-表示氯离子。参见ions.itp查看wrt力场中离子详细信息)
(3)-nn是指定加入的阴离子数目
(4)-g输出genion的log文件
(5)运行这个命令时,提示提供一个连续的溶剂组,应该是组12(SOL)。输入12,回车。程序会告知你有两个溶剂分子被氯离子代替
(6)添加离子的工具是genion模块。该模块会通读拓扑文件并以用户指定的离子代替水分子。
(7)输入文件即所谓的运行文件(run input file),文件后缀为.tpr;这个文件是通过grompp模块生成的,在后面运行第一次模拟的时候还会用到这个模块。
(8)在genion命令中,需要提供结构/状态文件(-s)作为输入文件,生成输出文件.gro (-o),处理拓扑文件(-p)反映替换掉的水分子和添加的离子,定义正负离子的名字(-pname和-nname),然后告诉genion添加中和体系电荷的正/负离子以及离子个数
(9)可以通过指定-neutral和-conc让genion添加固定浓度的离子而不是单单中和体系。可以参考genion的主页查看相关信息。-conc 0.15 浓度为0.15 M
(10)在之前的GROMACS版本中通过-pname和-nname指定的离子是力场规定的,但在4.5版本中则进行了标准化。规定的离子名称通常是大写的元素符号,并在[ molecules ]中命名然后写到拓扑文件中。根据不同的力场,残基名或者原子名可能带有+/-号,也可能不带。在genion命令中不要用原子或者残基名,否则在接下来的步骤中会遇到很多错误。
7.在进行动力学模拟之前,我们必须保证体系没有空间上的冲突或者不合理的构象。体系结构松弛是通过所谓的能量最小化(EM)实现的。
8.EM的过程与添加离子类似。我们再次利用grompp把体系结构、拓扑文件以及模拟参数集合到.tpr文件中,但是这次不是把.tpr文件传递给genion,而是通过mdrun运行能量最小化。
9.在运行genbox和genion时确保你已经更新了topol.top文件,否则会出现一大堆错误信息("number of coordinates in coordinate file does not match topology,“等)。
10.-v是给没有耐心等待的人设计的:它会让mdrun的输出变得冗长,每一步都会把过程输出到屏幕上。-deffnm用来定义输入和输出文件的名字。所以,如果你没有命名grompp输出文件"em.tpr”,你必须使用mdrun -s精确指定文件名。在本例中,我们会得到如下文件:
em.log:EM过程的ASCII码log文件
em.edr:二进制的能量文件
em.trr:二进制的全精度坐标文件
em.gro:能力最小化之后的结构
11.第一个是势能(potential energy,EM进程最后会输出)。Epot应该是负的,而且对于一个水中的简单的蛋白质体系来说处于105-106范围???,具体数值依赖于体系大小和水分子数目。第二个重要特征是最大作用力Fmax,收敛值是在minim.mdp中设置的——“emtol=1000.0”,意思是目标Fmax的值要低于1000KJ/(mol nm)。有时Fmax>emtol时Epot也可能处于合理范围,但是此时体系并没有完全稳定。此时应该评估为什么会出现这个现象,试着改变最小化参数(integrator、emstep等)。